 2016, Vol. 27
2016, Vol. 27 



In the research field of organic optoelectronics, π-functional molecules and polymers act as the great potential in the appliances of organic light-emitting diodes (OLEDs), organic photovoltaics (OPVs) and organic field-effect transistors (OFETs), due to their tunable optical and electrical properties owning to the majority of organic molecules and the variety of molecular stacking modes. The performance of the devices mentioned above strongly rely on the understanding of the behaviours of excitons, or the tight- binding electron-hole pairs to be exact, in organic materials, which is rather complex and different to that in the inorganic counterpart. The models on exciton were earlier developed by Frenkel and Davydov et al. [1], and recently were systematically discussed by Bredas and Spano et al. [2] through building more complex models. Radiative transition from excited molecules generates fluorescence when an electron relaxes from the excited singlet state to its ground state. However, the defects widely existing in most amorphous organic systems may quench fluorescence due to the trapping of excitons. Thus, the crystalline material usually presents the specific molecular stacking with very low content of impurities, making it precise to construct the relationship between the molecular stacking mode and the optoelectronic properties. The highly long-range ordered molecular arrangement induces the higher charge mobility [3], and at the same time, single crystal itself also could be highly efficient fluorescent material in solid [4].
There is still confusion on understanding the mobility of π- conjugated materials, but the consensus on designing of rigid structure with strong intermolecular interaction enables to achieve excellent performance [5]. According to the simplified Marcus theory which provides direct insight into the charge hopping between adjacent molecules, the charge transfer rate k is given by the following equation:
| $k=\left( \frac{4{{\pi }^{2}}}{h} \right){{t}^{2}}{{\left( 4\pi \lambda RT \right)}^{-0.5}}\exp \left( \frac{-\lambda }{4RT} \right)$ | (1) |
where λ is the internal reorganization term and t is transfer integral. Thus, smaller reorganization energy and larger transfer integral are of great benefit for the hopping rate. In fact, it is the stacking mode (the latter term), rather than intramolecular factor, that dramatically motivates changes in bandgap, especially results in a strong dispersion in the conduction band according to the one dimensional tight-binding model. A π-π distance of around 3.5 Å admittedly and 4 Å estimated in theoretical analysis implies the strong intermolecular electronic coupling, which consists with neighbouring molecules distance in most p stacking modes.
Herein, this short review will summarize how the stacking mode affects the optoelectronic functions of π-conjugated materials in the following three typical aggregation models including cofacial configuration (H-aggregation), staggered configuration (J-aggregation), and crossed configuration (X-aggre- gation), and probes into how to realize the balance of high solid state luminous efficiency and charge carrier mobility in one material.
2. The relationship between molecular stacking modes and optoelectronic functions of organic crystalline materialsAccording to molecular excitation model [6], the molecular excitation leads to transition dipole moment along the polarization axis. To a certain extent, the sign of the solvatochromism depends on the polarity in dipole moment of the molecule between its ground state and excited state. If the intermolecular overlap is large enough for units to abandon their individuality, it is well established that the primary photophysics in the bound dipoles, which is possibly coupled to a local lattice distortion of the conjugated cores in the orderly fashion [7]. The molecular stacking modes, indeed the relative spatial orientation of transition dipoles, do work in the energy splitting (so-called Davydov splitting) state of optically allowed transitions. The fluorescence usually originates from the decay of the lowest excited state, and as the result, H- and J-aggregation (parallel stacking mode) induce obvious spectral evolution while X-aggregation (cross stacking mode) keeps similar spectrum as single molecule, which is attributed to the spatial orientation of transition dipoles (Fig. 1). Kasha et al. concluded that the exciton state energy level diagrams are given by a succinct equation:
| $\Delta {{E}_{agg}}=\Delta {{E}_{mono}}+\Delta {{E}_{dv\pm \varepsilon }}$ | (2) |

|
Download:
|
| Figure 1. (a) Schematic representation of optically allowed transitions of molecular monomer and splitting in a cofacial configuration (H-aggregate), staggered configuration (J-aggregate) and crossed configuration (X-aggregation). (b) Splitting energy in the parallel and crossed stacking dimer systems in the framework of exciton model. | |
{kind=link}
where ΔEagg and ΔEmono represent for the upper energy levels of aggregate and monomer respectively; ΔEdv is the van der Waals term, which works prominently in the bound systems; and e is the splitting energy dependent on the dipoles moment, which is related to the centre-to-centre distance and relative spatial geometry between adjacent molecules. The orientation factor, κ, is usually used to describe the dimer geometry, actually in proportion to the splitting energy. In the dimer system, upon the parallel stacking mode, κ is given by κ|| = 1 -3 cos2 θ, which reflects that the upper state splitting for dimers with parallel transition moments varies the inclined angle θ. As a result, it could be optically distinguished through typical bathochromic shift of the J- aggregate band and hypsochromic shift of the H-aggregate band in the absorption spectra. Upon the crossed stacking mode, κ is given by κx = cosα (for the neglected intermolecular slippage, in other words, θ = 90°), which reflects that the upper state splitting for dimers with crossed transition moments varies the rotational angle α. As contrasted with parallel stacking mode, crossed stacking pattern exhibits simultaneously bathochromic and hypsochromic shift as the small rotational angle leads to both allowable splitting transition, while the large rotational angle makes the similar property of monomer. Thus in general, large intermolecular slippage (θ < 54.7°) and large intermolecular rotation are classified as J-aggregation and X-aggregation respectively, while the smaller slippage (θ > 54.7°) or smaller rotation is classified as H-aggregation. This classification is better understood and distinguished optically. Of cause, in the planar lattice system, the splitting energy increases as the aggregated length increases, which is corrected by a constant (relative to the number of interacting molecules), indicating the further narrowed bandgap of crystal.
On the other hand, the non-radiative transition is also considered in the fluorescence yield in solid, especially the long- range Forster energy transfer process. The structural factor, κ2, ranges from 0 to 4, is one of the crucial parameters to control the energy transfer efficiency (Fig. 2) [8]. The energy transfer rate κET is described as follows:

|
Download:
|
| Figure 2. κ2 in the parallel and crossed stacking modes and the inset shows the spatial orientation of energy donor and acceptor dipoles. In particular, orientation factor is given by κ2= (cos θT - 3 cos θD cos θA)2, where θT and θD(θA) are the angle between the donor and acceptor dipoles and the angle between the donor (acceptor) dipole and R (the vector from the donor to the acceptor), respectively. Adapted with permission from reference [8]. Copyright 2013 Royal Society of Chemistry. | |
{kind=link}
| ${{\kappa }_{ET}}=\frac{9000\left( 1n10 \right){{\kappa }^{2}}\varphi }{128{{\pi }^{5}}{{n}^{4}}{{N}_{A}}\tau {{R}^{6}}}Jc$ | (3) |
wherew φ is the quantum yield; n is the refractive index; NA is the Avogadro constant; τ is the lifetime and JC is the spectral overlap integral. Actually, there is always the case that the photoexcitaton agrees with the lowest allowable transition performs weak fluorescence, because J-aggregation (larger κ) is favoured for the energy transfer (non-radiative process). The effective energy transfer in long range might reflect the delocalization of excited species [9], and the intermolecular resonance might induce the loss of radiative probability. As has been remarked elsewhere [8, 10], if there is no obvious spectral shift for a polyatomic molecule in an aggregate, the rate of energy transfer may be quite significant. At the same time, κ2-derivational intermolecular Forster energy transfer was investigated in our group, by controlling dipoles of dopant to be embedded into the lattices without destroying the structural ordering. The parallel dipoles conduced to high transfer efficiency (κ2 = 1), and the perpendicular dipoles limited the transfer process (κ2 = 0), while the random dipoles were the intervenient one (κ2 = 2/3) that was common in amorphous film. As was reported in the doping systems (Fig. 3), DSB c Tc showed shorter Forster distance in crystal than in film, just similar to that for DSB c Pc due to the perpendicular orientation of dipoles in orderly structure. As for Ac c Tc, the parallel dipoles in crystal assisted for long-range Forster distance, relative to the random dipoles in film. Furthermore, a large-size (several millimetres) and ideal white-emissive (CIE: (0.33, 0.33)) organic crystal is obtained successfully by controllable energy transfer efficiency.

|
Download:
|
| Figure 3. Schematics of relative spatial dipoles in the doping crystal systems: (a) DSB c Tc crystal (κ2 = 0); (b) DSB c Tc film(κ2 = 2/3); (c) Ac c Tc crystal (κ2 = 1); and the red axis represents the corresponding molecular dipole direction. Adapted with permission from reference [8]. Copyright 2013 Royal Society of Chemistry. | |
{kind=link}
2.1. Cofacialconfiguration (H-aggregation)
It is a common phenomenon for π-conjugated molecules to spontaneous aggregate into orderly fashion due to their planar conformation and strong contact along π-π direction. Thus the cofacial configuration widely exists in most systems, especially π-scaffolds of acenes, thienoacenes, and dye molecules of cyanines, porphyrins, perylenebisimides etc. There is no doubt that excellent charge mobility could be obtained in this stacking pattern, such as rubrene single crystal with high record of 43 cm2 V-1 s-1 and pentacene crystalline thin film of 40 cm2 V-1 s-1 [5]. A great amount of effort is paid on tuning the stacking mode by introducing heteroatoms, outside strain, steric effect etc. The dense packing and π-orbits overlap offer the maximum transfer integral, especially as for the two dimensional cofacial stacking mode, contributing to the higher mobility. Theoretically, the graphenelike molecules could perform the expected charge mobility because of their ideal two-dimensional growth into lamellar structure with large π-overlap. However, the difficulties of processing for H-type molecule are their poor solubility due to the strong π-π interaction. Simply but directly, long alkyl-chain modification and/or finite twisted configuration is introduced to most of molecular design, but in nature, the weakened π-π interaction or unexpected stacking mode seems to comprise on the solubility. Interestingly, as shown in Fig. 4, discotichexa-peri- hexabenzocoronene (HBC), which was regarded as the smallest graphene fragment, was in situ electrochemical synthesized by soluble hexaphenylbenzene (HPB) and directly deposited onto the electrode with a discrete nanofibular structure. The large π-πlane tended to be face-to-face stacking into mole cular column, which is beneficial for the charging/discharging process. The strategy of electrochemical synthesis and in situ assembly of large, planar hydrocarbons constitutes an important step towards applications of insoluble π-conjugated molecules [11].

|
Download:
|
| Figure 4. Schematic of in situ electrochemical synthesis and self-assembly of discotic HBC. Adapted with permission from reference [11]. Copyright 2013 Wiley-VCH Verlag GmbH & Co. | |
{kind=link}
Actually, it seems to be possible to achieve the fluorescent H-type molecules, although strictly speaking it turns to not be the cofacial stacking pattern. (i) The so-called herringbone stacking mode could not be classified as the H- or J-aggregation, since the orientation of adjacent dipoles with a cell unit form an oblique angle. In principle, there exist the in-phase and out-ofphase arrangements of transition dipoles. Such kind of stacking pattern simultaneously possesses the H-type and J-type characters, thus leads to the spectral red and blue shifts in the absorption and only the red shift in the fluorescence due to strong excitonic coupling (see the detail in J-aggregation). On the other hand, this stacking mode makes it possible to be densely packed into a planar crystal with higher mobility and fluorescence quantum yield, even achieving superradiance in H- aggregates [12]. (ii) Face-to-face stacking mode with small rotational angle is also favoured to promoting fluorescence. The ideal cofacial configuration is a quenching emitter system due to the lowest splitting energy state is forbidden, while the rotational angle induces the optically allowable process. The finitely rotational H-like aggregation naturally retains more H- type property, more or less, with relatively compromising photoluminescence quantum yield [13]. (iii) H-aggregate may lead to the enhancement of quantum yield of phosphorescence at the expense of fluorescence [14]. The cofacial configuration is predicted as the maximum enhancement of phosphorescence since the singlet excitons in the upper state is forbidden to the fluorescence transition and the comparatively well-matched energy to realize conversion. More importantly, the relative rates of intersystem crossing (singlet-triplet internal conversion) may determine the final property, although the order of magnitude of aggregation singlet-splitting (1000 cm-1) is comparable with the lowest singlet -triplet split for dye monomers (1000-3000 cm-1). (iv) The fluorescent H-aggregate might be obtained through S2-S0 pathway. Basak et al. [15] reported NDI aggregates in a binary (good and poor) solution, which was observed a broad excimer emission, and they attributed these rare phenomena as the allowable excitonic transition of S2-S0 due to the very similar dimer configuration of S2 and S1 and expatiated that the competing internal conversion (S2-S1) was a slow process relative to intense emission from the high lying excited state (S2-S0).
On the other hand, although the splitting upper state shows the lowest forbidden transition as the result of which most of energy loss is the non-radiative process such as the long-range energy transport, it is favoured for achieving a stable charge-separated state [16]. The non-radiative loss means the most effective power conversion, which shows great potential in the field of photodetectors and biosensors. Andreas etal. reported a one dimensional self-assembly of carbonyl-bridged triarylamin (CBT), which was aligned cofacially to form supramolecular building blocks. The orderly CBTs assisted for delocalization of electric excitations and coherent dominated energy transport over long distance (several micrometres at room temperature).
2.2. Staggered configuration (J-aggregation)Nowadays, J-aggregates are still of significant interest due to their charming characters, such as high fluorescence intensity with small Stokes shift and exceptional capacity of exciton delocalization and transport. The most attractive phenomena are the so- called superradiance [17], the cooperative emission property due to their lowest excitonic transition. At the ultralow temperature, the fluorescent lifetime of J-aggregates is decreasing much shorter than that of monomers, which indicates a large number of coherently coupled molecules and thus long-range excitons delocalization. The higher excitation density and consistent dipoles orientation (or largely parallel to each other) is propitious to the accumulated photon echo. In principle, one dimensional stacking mode of J-aggregates in an orderly fashion is favoured for the high fluorescence.
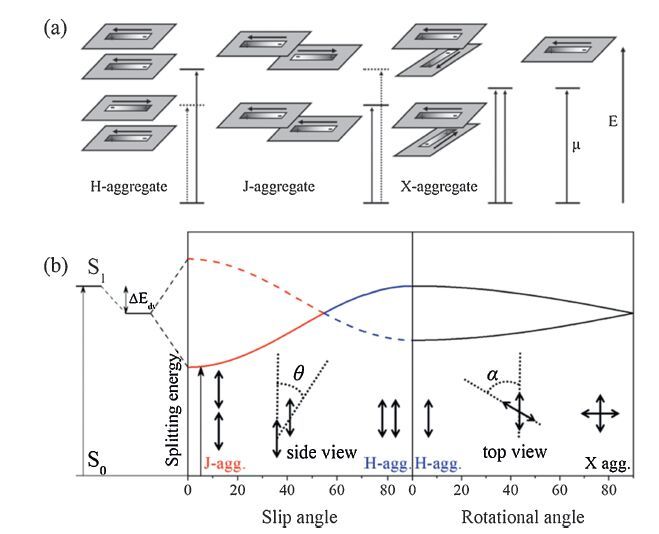
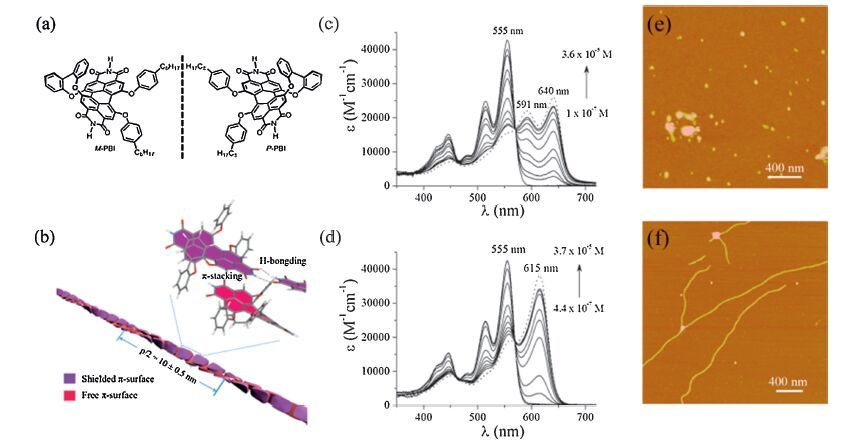
In general, H-type to J-type stacking transformation is efficient by introducing spatial groups for finite slipped π-conjugated cores [18]. Recently, a novel asymmetric and twistedperylenebisimide (PBI) was designed to construct J-aggregates by introducing a biphenoxy bridge at one of the bay areas and two flexible phynoxy groups at the opposite side. The M and P atropo-enantiomers were successfully isolated at room temperature due to the increased rigidity of the molecular backbone owing to the bridged structure (Fig. 5) [19]. The resulting experiments showed that the enantiopureperylenebisimide (M-PBI and P-PBI) could well form extended one-dimensional nanowires with J-type excitonic coupling due to the mutual effects of hydrogen bonding and π-π interaction. The highly orderly homochiral units tended to be organized to nanowires and revealed the high photoluminescence efficiency of 47 ± 3%. On the other hand, for the racemate (rac-PBI), an enantiomer preferentially recognized its mirror image, that is, the dominated self-discrimination inhibited the continuous extension and resulted in the formation of zero-dimensional particle-like aggregates. As the case stands, the absorption spectra reflected one intense J-band at 615 nm for M-PBI (or P-PBI), but two distinctive J-type bands for rac-PBI (M-PBI and P-PBI). The fluorescence spectra of the aggregates of M-PBI(or P-PBI) and rac-PBI showed respectively pronounced red shifts of 45 nm and 70 nm relative to the monomers. Moreover, photoluminescence efficiency of particle-like aggregates revealed only 12 ±3%, much smaller than highly ordered one (47 ± 3%). The difference of fluorescent characters absolutely depends on the stacking pattern, which the orderly fashion ensures the long-range migration of excitons (similar to the superradiative performance), while the racemic mixture act as the traps of each other, leading to the short-lived excitons. This discovery could explain the efficient fluorescence quenching of more concentrated solution (the so-called superquenching), which wildly exists in most conjugated system. One dimensional assembly of J-aggregate is always to be distorted as aggregate length increases, indicating the defects are easily formed even though the intensive supramolecular force drives the units to preferentially grow. Thus the atactic configuration indeed works on quenching, originated from the trapping of excitons.

|
Download:
|
| Figure 5. (a) Chemical structures of atropo-enantiomericperylenebisimidesand M-PBI and P-PBI; (b) schematic representation for the aggregate of the enantiopure PBI (M-PBI); (c) and (d) concentration-dependent UV/vis absorption spectra of M-PBI (or P-PBI) and rac-PBI (the dotted lines show the calculated absorption spectra of the aggregates of M- PBI and rac-PBI, respectively), and the inset pictures show respective AFM morphology. Adapted with permission from reference [19]. Copyright 2012 Wiley-VCH Verlag GmbH & Co. | |
{kind=link}
However, long linear or branched alkyl chain is always ineluctable to improve the solubility as the expense of π-π interaction and capacity of crystallization. In general, introducing the spatial groups, even the methyl group in the proper position could make a difference to stacking pattern. Recently, a novel PBI derivative has been reported that lacks any flexible solubilizing groups and demonstrate the self-assemble into three dimensional nanofibroid networks dispersed in solution and even in polymer matrices [20]. The concept for molecular design was based on π-scaffold contortion with a small methyl group, which led to the great difference in the solubility and self-assembly capacity (from H-aggregation to J-aggregation). The needle-like single crystal with a width of about ten micrometres could be easily prepared. The J-aggregates exhibited a high fluorescence quantum yield of 0.77 ± 0.03. Moreover, in order to take full advantage of long-range exciton migration, this J-aggregate had been successfully applied as cathode interlayer materials in highly efficient inverted polymer solar cells [21]. The rod-like aggregates with J-type excitonic coupling were self-assembly formation by spin-coating method from anhydrous THF. The enhanced power conversion efficiency was up to >9% with a conventional structure of ITO/PBI-H(10 nm)/PTB7:PC71BM/MoO3/Al, which was attributed to expected electron mobility in their highly ordered nanostructure and matched energy level.
Indeed, when considering the intermolecular π-orbital overlap the staggered configuration is not an ideal strategy for designing on high charge mobility due to the displacement of adjacent molecules. Ribierre et al. [22] reported a π-type bisazomethine dye thin film with poor mobility of around 10_4 cm2 V_1 s_1 in the bottom-gate toπ-contact OFETs. However, as mentioned before, HJ-aggregates could show the excellent charge mobility, as well as the fluorescent property. The positive (negative) H- and negative (positive) J-type induced the ratio of the first two vibronic peak intensities in the absorption and fluorescence spectra was investigated by Spano [2, 23], indicating that in a unit cell, oscillator strength generally occurred in the top and bottom of the exciton band, leading to optical properties that contained elements of both ideal J- and H-aggregate behaviours. These so-called herringbone stacking modes are common in acenes [24], oligo(π-πhenylenevinylene) derivatives [25], phenylene/thioph- ene co-oligomers [26], which could balance high fluorescence quantum yield and charge mobility in crystalline state. For example, Deng et al. [27] reported an angular slice-like crystal of cyano-substituted oligo(π-πhenylenevinylene), which showed the photoluminescence quantum yield of 75% with intensive self- waveguided emission. Moreover, ambipolar characters of single crystal FET with the symmetric source/drain (Au) electrodes was observed, exhibiting high and balanced electron and hole mobility of 0.745 cm2 V-1 s-1 and 0.239 cm2 V-1 s-1. With the asymmetric source/drain (Au and Ca) electrodes, the ambipolar mobility was as high as 2.50 cm2 V-1 s-1 and 2.10 cm2 V-1 s-1 for electron and hole respectively. Deservedly, such kinds of molecules are the promising materials to realizing the laser diode, which ought to be tolerant of high charge injection and efficient conversion of fluorescence.
2.3. Crossed configuration (X-aggregation)The concept of X-aggregation to enhance fluorescence was put forward theoretically by Cornil etal. [28] in 1998. As the rotational angle of dimer increases, the energy splitting excited states both realize the optically allowable transitions. Specially, when the rotational angle increases up to 90°, the dipole-dipole coupling is neglectable, and the exciton is localized within several units due to the special structural factor (zero) and the limited energy transfer rate. In such a stacking pattern with perpendicularly orientated dipoles, the shrinkage of dipole interaction will lead to an intense absorption and fluorescence similar to the monomeric molecule.
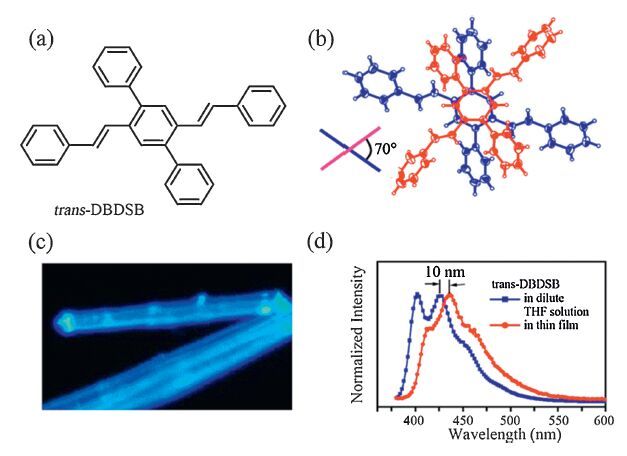
Although X-aggregation was predicted for a long time to be the promising way to prevent the fluorescence quenching, the first experimental observation in the needle-like crystal of 2, 5- diphenyl-1, 4-distyrylbenzene (trans-DPDSB) was reported by our group in 2005 [29]. This is because the crossed configuration shows the higher ground state energy in a thermodynamic instability. In this case, PPV-type oligomer seemed to be parallel stacking along its long molecular axis, but trans-DPDSB was creatively introduced with two phenyl substituents to the central phenyl ring, which led to the CH/p intermolecular interactions, as well as the stereospecific blockade to form the cross-stacking mode (Fig. 6). As the result, the molecules were packed into one- dimension column along b axis, with relatively rotational angle of 700°, and the distance between the central rings of adjacent molecules was 4.1 Å (b/2), slightly larger than the conventional π-π contact. The attractive feature was that the PL spectra of trans-DPDSB were quite similar in dilute THF solution and solid film. Although it is difficult to achieve the ideal perpendicular configuration, the fluorescence quantum yield in crystalline state was still on top at 0.8, and 10 nm red shift was observed in the PL spectra from the molecularly dissolved solution to the aggregated thin film. Additionally, a series of electroluminescent devices suggested the excellent electron transporting property. The EL spectra also exhibited the pure blue emission with device structure of ITO/PEDOT:PSS/NPB(50 nm)/trans-DPDSB(20 nm)/ Alq(30 nm)/Al, where PEDOT:PSS, NPB and Alq acted as hole injecting layer, hole transporting layer and electron transporting layer respectively.

|
Download:
|
| Figure 6. (a)Chemicalstructureof trans-DPDSB;(b)crossedstackingdimerof trans-DPDSBwiththerotationalangleof700;(c)singlecrystalunderUVlamp; (d) fluorescence spectra of trans-DPDSB in dilute THF solution and thin film. Adapted with permission from reference [29]. Copyright 2005 American Chemical Society. | |
{kind=link}
In most cases, the molecule with rigid and cruciform structure is more likely to pack as X-aggregation. Generally speaking, most of crossed stacking molecules are based on the introduction of groups at the side positions perpendicular to long molecular axis, such as the PPV and PPE oligomers. The resulting intermolecular interaction, such as the H bonds, CH/π, becomes the key driving force to form the crossed configuration. In 2006, Sanyal et al. [30] designed and obtained the crossed dipole π-stacking 1, 4-bis(phenylethy- nyl)benzene (BHE-PE2.5). The H-type PE2.5 was no doubt packed along the c axis, while the BHE groups assisted to form hydrogen bonds and drive the rotational configuration. Another example is that the different optical properties of 2B2N-DSB and 2B2N-DEB. Compared with the stacking mode in spin-coated film, 2B2N-DSB was parallel stacking while the 2B2N-DEB was crossed one, and as the result, the photoluminescence quantum yield of former was 73%, lower than that of the latter one (90%) [30]. Ma et al. [31] also reported a crossed stacking molecule 1, 4-bis(2, 2-diphenylvinyl)- benzene (BDPVB), which showed the aggregation-induced emission (AIE) property with a low fluorescence quantum yield of 5% in dilute THF solution and 50% in crystalline state.
In view of fluorescence, the crucial parameter of X-aggregation is the rotational angle between the adjacent molecules (the relative position of dipole orientation indeed). For example, the rotational angle of trans-DSB, trans-DPDMeSB and trans-DPDSB is 00°, 250°, and 700°, and the fluorescence quantum yield is <0.1, 0.62 and 0.8, respectively [32]. From another aspect, the profound guide to realize the fluorescent H-type molecule is to induce the fliπ-flap geometry. Che et al. [33] found the symmetric CH-PTCDI with highly efficient fluorescence quantum yield of 17%, which was great leap in such bound system (typically <1% in these H-type dye molecules), just due to the slightly rotational angle of 400°. There is no doubt that the lar ger rotational angle could dramatically improve the fluorescent property, even the little angle could make a great difference.
On the other hand, the charge transporting property of X- aggregation is hardly investigated due to the large π-π distance. Xiao et al. [34] reported a series of pentacenederivertives that are substituted along their edges with aromatic rings. Among their crystalline states, 6, 13-diphenylpentacenetended to assemble into X-type stacking mode, and 6, 13-di(20-thienyl)pentacenetended to assemble into H-type stacking mode, in which the driving forces are CH/p and CH/S interaction respectively. The former showed the poor mobility of 8×10-5 cm2 V-1 s-1, while the latter showed the comparatively good mobility of 0.1 cm2 V-1 s-1 in thin film transistors, which revealed that the crossed stacking mode may result in much traps due to the larger intermolecular distance. Liu et al. [35] designed a TES-DPA, which was packed along the c axis to form the one dimension molecular column with the rotational angle of 600. It showed a solid state fluorescence yield of 77.3%. The charge carrier mobility of 1.47 cm2 V-1 s-1was measured by the single crystal FET device, integrating optical and electrical properties.
3. Summary and perspectiveIn this short review, we have analyzed the relationship between the stacking mode and the optoelectronic functions of π- conjugated materials. The aggregate state is no doubt that influence the energy band level by the coupling π-orbits, leading to the spectral shift and electric property. The crystalline material tends to construct the structure-performance relationship, and on the other hand, the low content of impurity ensures the highly efficient optoelectric properties because the defects could easily trap the excitons and charge carries. Moreover, energy transfer process could not be neglected because it might be one of the key factors on the degree of excitonic delocalization. Considering on the combination ofhighly optical and electric property, J-aggregate with high mobility or H-aggregate with high fluorescence are both feasible ways, more or less, which have been reflected in recent work. The parallel stacking mode betwixt H- and J-aggregation may provide a novel approach towards highly optical and electric property by integrating the respective advantages of H- and J-aggregation.
Acknowledgments We thank the financial support from the National Natural Science Foundation of China (Nos. 51373054, 51573055, 51473052, 51521002, 21334002), National Basic Research Programme of China (973 Programme, Nos. 2013CB834705, 2014CB643504), Fundamental Research Funds for the Central Universities and Introduced Innovative R&D Team of Guangdong (No. 201101C0105067115)| [1] | A.S. Davydov. Theory of absorption spectra of molecular crystals. Ukr. J. Phys. 53 (2008) 65–70. |
| [2] | (a) S.V. Frolov, W. Gellermann, Z.V. Vardeny, M. Ozaki, K. Yoshino, Picosecond photophysics of luminescent conducting polymers from excitons to polaron pairs, Synth. Met. 84(1997) 493-496; (b) J. Cornil, D. Beljonne, J.P. Calbert, J.L. Brédas, Interchain interactions in organic π-conjugated materials: impact on electronic structure, optical response, and charge transport, Adv. Mater. 13(2001) 1053-1067; (c) V. Coropceanu, J. Cornil, D.A. da Silva Filho, et al., Charge transport in organic semiconductors, Chem. Rev. 107(2007) 926-952; (d) F.C. Spano, The spectral signatures of Frenkelpolarons in H-and J-aggregates, Acc. Chem. Res. 43(2009) 429-439. |
| [3] | M.E. Gershenson, V. Podzorov, A.F. Morpurgo. Colloquium:electronictransport in single-crystal organic transistors. Rev. Mod. Phys. 78 (2006) 973–989. DOI:10.1103/RevModPhys.78.973 |
| [4] | M. Shimizu, T. Hiyama. Organic fluorophores exhibiting highly efficient photoluminescence in the solid state. Chem. Asian J. 5 (2010) 1516–1531. DOI:10.1002/asia.v5:7 |
| [5] | H.L. Dong, X.L. Fu, J. Liu, Z.R. Wang, W.P. Hu. 25th anniversary article: key points for high-mobility organic field-effect transistors. Adv. Mater 25 (2013) 6158–6183. DOI:10.1002/adma.v25.43 |
| [6] | M. Kasha, H.R. Rawls, M.A. El-Bayoumi. The exciton model in molecular spectroscopy. Pure Appl. Chem. 11 (1965) 371–392. |
| [7] | R.M. Hochstrasser, M. Kasha. Application of the exciton model to mono-molecular lamellar systems. Photochem. Photobiol. 3 (1964) 317–331. DOI:10.1111/php.1964.3.issue-4 |
| [8] | H. Wang, B.L. Yue, Z.Q. Xie, et al. Controlled transition dipole alignment of energy donor and energy acceptor molecules in doped organic crystals, and the effect on intermolecular Förster energy transfer, Phys. Chem. Chem. Phys. 15 (2013) 3527–3534. DOI:10.1039/c3cp43800g |
| [9] | M. Kasha. Energy transfer mechanisms and the molecular exciton model for molecular aggregates. Radiat. Res. 178 (2012) AV27–AV34. DOI:10.1667/RRAV03.1 |
| [10] | O.V. Mikhnenko, P.W.M. Blom, T.Q. Nguyen. Exciton diffusion in organic semiconductors. Energy Environ. Sci. 8 (2015) 1867–1888. DOI:10.1039/C5EE00925A |
| [11] | L.Q. Qin, Y.N. Zhang, X.Y. Wu, et al. In situ electrochemical synthesis and deposition of discotichexa-peri-hexabenzocoronene molecules on electrodes: self-assembled structure, redox properties, and application for supercapacitor. Small 11 (2015) 3028–3034. DOI:10.1002/smll.v11.25 |
| [12] | (a) F. Meinardi, M. Cerminara, A. Sassella, R. Bonifacio, R. Tubino, Superradiance in molecular H aggregates, Phys. Rev. Lett. 91(2003) 247401; (b) S.H. Lim, T.G. Bjorklund, F.C. Spano, C.J. Bardeen, Exciton delocalization and superradiance in tetracene thin films and nanoaggregates, Phys. Rev. Lett. 92(2004) 107402. |
| [13] | (a) F. Würthner, Z.J. Chen,V. Dehm, V. Stepanenko, One-dimensional luminescent nanoaggregates of perylenebisimides, Chem. Commun. (2006) 1188-1190; (b) Z.J. Chen, V. Stepanenko, V. Dehm, et al., Photoluminescence and conductivity of self-assembled π-π stacks of perylenebisimide dyes, Chem. Eur. J. 13(2007) 436-449. |
| [14] | E.G. McRae, M. Kasha. Enhancement of phosphorescence ability upon aggregation of dye molecules. J. Chem. Phys. 28 (1958) 721–722. DOI:10.1063/1.1744225 |
| [15] | S. Basak, N. Nandi, K. Bhattacharyya, A. Datta, A. Banerjee. Fluorescence from an H-aggregated naphthalenediimide based peptide: photophysical and computational investigation of this rare phenomenon. Phys. Chem. Chem. Phys. 17 (2015) 30398–30403. DOI:10.1039/C5CP05236J |
| [16] | A.T. Haedler, K. Kreger, A. Issac, et al. Long-range energy transport in single supramolecularnanofibres at room temperature. Nature 523 (2015) 196–199. DOI:10.1038/nature14570 |
| [17] | F.C. Spano, S. Mukamel. Superradiance in molecular aggregates. J. Chem. Phys. 91 (1989) 683–700. DOI:10.1063/1.457174 |
| [18] | S. Ghosh, X.Q. Li, V. Stepanenko, F. Würthner. Control of H-and J-type π stacking by peripheral alkyl chains and self-sorting phenomena in perylenebisimide homo-and heteroaggregates. Chem. Eur. J. 14 (2008) 11343–11357. DOI:10.1002/chem.200801454 |
| [19] | (a) Z.Q. Xie, F. Würthner, Perylenebisimides with rigid 2,2'-biphenol bridges at bay area as conjugated chiral platforms, Org. Lett. 12(2010) 3204-3207; (b) Z.Q. Xie, V. Stepanenko, K. Radacki, F. Würthner, Chiral J-aggregates of atropoenantiomericperylenebisimides and their self-sorting behavior, Chem. Eur. J. 18(2012) 7060-7070. |
| [20] | Z.Q. Xie, V. Stepanenko, B. Fimmel, F. Würthner. An organogelator design without solubilizing side chains by backbone contortion of a perylenebisimide pigment. Mater. Horiz. 1 (2014) 355–359. DOI:10.1039/c3mh00159h |
| [21] | Z.Q. Xie, B. Xiao, Z.C. He, et al. Self-assembled perylenebisimide J-aggregates as promising cathode modifiers for highly efficient inverted polymer solar cells. Mater. Horiz. 2 (2015) 514–518. DOI:10.1039/C5MH00056D |
| [22] | J.C. Ribierre, M. Sato, A. Ishizuka, et al. Organic field-effect transistors based on J-aggregate thin films of a bisazomethine dye. Org. Electron. 13 (2012) 999–1003. DOI:10.1016/j.orgel.2012.02.020 |
| [23] | F.C. Spano, C. Silva. H-and J-aggregate behavior in polymeric semiconductors. Annu. Rev. Phys. Chem. 65 (2014) 477–500. DOI:10.1146/annurev-physchem-040513-103639 |
| [24] | (a) J.E. Anthony, Functionalized acenes and heteroacenes for organic electronics, Chem. Rev. 106(2006) 5028-5048; (b) J.E. Anthony, The larger acenes: versatile organic semiconductors, Angew. Chem. Int. Ed. 47(2008) 452-483. |
| [25] | J. Gierschner, S.Y. Park. Luminescent distyrylbenzenes: tailoring molecular structure and crystalline morphology. J. Mater. Chem. C 1 (2013) 5818–5832. DOI:10.1039/c3tc31062k |
| [26] | M.H. Yoon, A. Facchetti, C.E. Stern, T.J. Marks. Fluorocarbon-modified organic semiconductors: molecular architecture, electronic, and crystal structure tuning of arene-versus fluoroarene-thiophene oligomer thin-film properties. J. Am. Chem. Soc. 128 (2006) 5792–5801. DOI:10.1021/ja060016a |
| [27] | (a) J. Deng, J. Tang, Y.X. Xu, et al., Cyano-substituted oligo(p-phenylenevinylene) single-crystal with balanced hole and electron injection and transport for ambipolar field-effect transistors, Phys. Chem. Chem. Phys. 17(2015) 3421-3425; (b) J. Deng, Y.X. Xu, L.Q. Liu, et al., An ambipolar organic field-effect transistor basedonanAIE-activesinglecrystalwithahighmobilitylevelof 2.0cm2V-1s-1, Chem. Commun. 52(2016) 2370-2373. |
| [28] | (a) J. Cornil, D.A. Dos Santos, X. Crispin, R. Silbey, J.L. Brédas, Influence of interchain interactions on the absorption and luminescence of conjugated oligomers and polymers: a quantum-chemical characterization, J. Am. Chem. Soc. 120(1998) 1289-1299; (b) M.C.R. Delgado, K.R. Pigg, D.A. da Silva Filho, et al., Impact of perfluorination on the charge-transport parameters of oligoacene crystals, J. Am. Chem. Soc. 131(2009) 1502-1512. |
| [29] | Z.Q. Xie, B. Yang, F. Li, et al. Cross dipole stacking in the crystal of distyrylbenzene derivative: the approach toward high solid-state luminescence efficiency. J. Am. Chem. Soc. 127 (2005) 14152–14153. DOI:10.1021/ja054661d |
| [30] | (a) N. Sanyal, P.M. Lahti, Hydrogen-bond-assisted, crossed dipole π-stacking in 1, 4-bis (phenylethynyl) benzene, Cryst. Growth Des. 6(2006) 1253-1255; (b) C.H. Zhao, A. Wakamiya, Y. Inukai, S. Yamaguchi, Highly emissive organic solids containing 2,5-diboryl-1,4-phenylene unit, J. Am. Chem. Soc. 128(2006) 15934-15935. |
| [31] | S.Q. Ma, J.B. Zhang, J.Y. Qian, et al. Efficient spontaneous and stimulated emission from 1,4-bis(2,2-diphenylvinyl)benzene single crystals with cross-dipole stacking. Adv. Opt. Mater. 3 (2015) 763–768. DOI:10.1002/adom.v3.6 |
| [32] | (a) Z.Q. Xie, W.J. Xie, F. Li, et al., Controlling supramolecular microstructure to realize highly efficient nondoped deep blue organic light-emitting devices: the role of diphenyl substituents in distyrylbenzene derivatives, J. Phys. Chem. C 112(2008) 9066-9071; (b) B. Yang, Y.G. Ma, J.C. Shen, Stacking mode, optoelectronic property and supramolecular control method in π-conjugated organic molecules, Chem. J. Chin Univ. 29(2008) 2643-2658. |
| [33] | Y.K. Che, X.M. Yang, K. Balakrishnan, J.M. Zuo, L. Zang. Highly polarized and selfwaveguided emission from single-crystalline organic nanobelts. Chem. Mater. 21 (2009) 2930–2934. DOI:10.1021/cm9007409 |
| [34] | Q. Miao, X.L. Chi, S.X. Xiao, et al. Organization of acenes with a cruciform assembly motif. J. Am. Chem. Soc. 128 (2006) 1340–1345. DOI:10.1021/ja0570786 |
| [35] | J. Liu, L.Q. Meng, W.G. Zhu, et al. A cross-dipole stacking molecule of an anthracene derivative: integrating optical and electrical properties. J. Mater. Chem. C 3 (2015) 3068–3071. DOI:10.1039/C4TC02964J |