 2016, Vol. 27
2016, Vol. 27 


Currently, hepatitis C virus (HCV) infection has become a major public health problem and heavy burden worldwide [1-3]. Recently, the direct-acting antivirals (DAAs), which specifically target HCV proteins had made great progresses. Telaprevir and Boceprevir in combination with peg IFN-a plus RBV, as the first generation of NS3/4A HCV protease inhibitors, have been widely used in clinic for the treatment of HCV infections. However the side effects, drug interactions and resistance mutations have limited their clinical use [4-7]. Several new HCV DAAs and their combinations have been approved since the end of 2013, such as NS3/4A inhibitors Simeprevir and Asunaprevir, NS5A inhibitors Ledipasvir and Daclatasvir and NS5B polymerase inhibitors Sofosbuvir and Dasabuvir [8]. Especially, some new HCV DAAs and their triple therapies show significant promise in some drugresistant HCV in clinical patients. However, their chemotherapeutic pressure is on viral components; thus, antiviral therapies targeting specific viral enzyme might cause drug-resistant mutations [9]. Therefore, novel antiviral therapeutic strategies against HCV with no or decreased chance of inducing drugresistance are highly desirable [10].
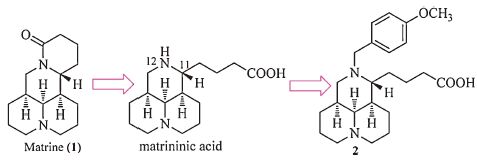
We have successfully identified 12-benzyl matrinic acid analogs to be a novel class of anti-HCV agents from matrine (1, Fig. 1), a Chinese natural product [11]. The representative compound, 12-p-methoxylbenzyl matrinic acid (2, Fig), demonstrated a moderate activity with EC50 of 118 mmol/L and SI of over 22 [12-14]. Its mode of action is down-regulating host heat-stress cognate 70 (Hsc70) protein expression at the post transcriptional level through destabilizing Hsc70 mRNA, a distinctly different action mode from the currently used drugs such as Telaprevir and Simeprevir [12]. As the target is not on the viral component, this kind of antiviral agents might inhibit viral replication with decreased chance of causing drug-resistant mutations [14].

|
Download:
|
| Figure 1. Chemical structures of matrinine (1) , matrinic acid and 12-p-methoxylbenzyl matrinic acid (2) . | |
This unique action mode and special scaffold of compound 2 strongly provoked our interest to continuously explore the structure-activity relationship (SAR) of this kind of compounds, in an effort to discover novel anti-HCV candidates with an advantage of overcoming the drug resistance. Therefore, in the present study, taking compound 2 as the lead, SAR studies were further conducted with the variations of the 40-carboxyl group and the shortening length of the 11-side chain. Based on this strategy, a series of new 12-benzyl matrinic amide/ethanamide and matrinic acid/amine derivatives were constructed and evaluated for their in vitro anti-HCV activity as well as the in vivo pharmacokinetic (PK) and safety profile of the representative compounds.
2. Experimental 2.1. ChemistryMelting points (mp) were obtained using a CXM-300 melting point apparatus. 1 H NMR and 13C NMR spectra were recorded on a Bruker Avance 400 spectrometer or Bruker Avance Ⅲ 500 spectrometer (Varian, San Francisco, CA) respectively, in CDCl3, DMSO or CD3OD using Me4Si as the internal standard. Flash chromatography was performed on Combiflash Rf 200 (Teledyne, Nebraska, USA). All the target compounds were synthesized using commercially available sophocarpine (3) and matrine (1) with purity over 98% as the starting material, which were purchased from the Yanchi Dushun Biological and Chemical Co., Ltd. (Shanxi, China).
2.1.1. General procedures for methyl N-substituted matrinic acetate (16a-b)Matrine (1, 1.0 g, 4 mmol) was added to 5 mol/L NaOH (20 mL), and the reaction mixture was heated under reflux for 9 h, cooled in an ice bath and then acidified with HCl (6 mol/L) to pH 4-5. The solvent was removed under reduced pressure, and the residue was dissolved in methanol (100 mL). The suspension was filtered and the filtrate was concentrated. The residue was dissolved in 2 mol/L HCl/MeOH (20 mL) and the mixture was refluxed for 2 h. The crude compound 15 was obtained by evaporation and used in the next step without further purification. Anhydrous K2CO3 (3.5 equiv.) and benzyl bromide (1.5 equiv.) were added to a solution of compound 15 (1.0 equiv.) in MeCN (20 mL), and the reaction solution was stirred at room temperature until TLC analysis showed completion of the reaction. The reaction mixture was filtered, and the filtrate was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 and washed by water and brine, dried with anhydrous Na2SO4, filtered, and concentrated to afford crude compounds 16a-b. The title compounds 16a-b were obtained by purifying with flash column chromatography on silica gel with dichloromethane and methanol as the eluents in yields of 50%-60%.
2.1.2. 12-(4-Pyridylmethyl) matrinic acid dihydrochloride (17)Compound 16b (1.5 mmol) was refluxed in 3 mol/L HCl (15 mL) for 1 h, then cooled, and adjusted to pH 5-6 with 3 mol/L KOH. The solution was extracted with ethyl acetate, and the aqueous layer was evaporated to dryness, and the residue was purified through flash chromatography over silica gel to give the title compounds 17. White solid yield: 83%, mp: 175-177 ℃; 1H NMR (400 MHz, DMSO-d6): 12.16 (s, 1H), 12.01 (s, 1H), 11.08 (s, 1H), 8.68 (d, 2H, J = 6.0) , 7.73 (d, 2H, J = 6.0) , 5.02 (s, 1H), 4.22 (s, 1H), 4.05-3.95 (m, 2H), 3.58 (s, 1H), 3.24 (t, 2H, J = 14.0) , 2.84-2.96 (m, 2H), 2.78 (s, 1H), 2.65 (s, 1H), 2.50 (s, 1H), 2.34 (s, 2H), 1.51-2.07 (m, 12H); 13C NMR (101 MHz DMSO-d6): 174.3, 149.5 (2) , 139.4, 126.0 (2) , 60.7, 60.2, 56.0, 54.2 (2) , 49.1, 35.9, 32.8, 29.9, 27.9, 23.9, 23.5, 21.6, 17.9 (2) ; HRMS: calcd. for C21H32N3O2-2HCl [M-2HCl+H]+: 358.2495, found: 358.2482.
General procedures for other title compounds and their physical and spectroscopic characterization data were given in Supporting information.
2.2. Biological methodsHuman liver cell line Huh7.5 cells (kindly provided by Vertex Pharmaceuticals, Inc., Boston, MA) were cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% inactivated fetal bovine serum and 1% penicillin-streptomycin (Invitrogen). Cells were digested with 0.05% trypsin-ethylene diamine tetraacetic acid (EDTA) and split twice a week.
2.2.1. Anti-HCV effect in vitro [15]Huh7.5 cells were seeded into 96-well or 6-well plates (Costar) at a density of 3.0 × 104 cells/cm2. After 24 h of incubation, the cells were infected with HCV viral stock (recombination virus strain J6/JFH/JC, 45 IU/cell) and simultaneously treated with the test compounds at various concentrations. The culture medium was removed after 72 h of incubation, and the intracellular total RNA (in 96-well plates) was extracted with RNeasy Mini Kit (Qiagen), and total intracellular proteins (in 6-well plates) were extracted with Cyto-Buster Protein Extraction Reagent added with 1 mmol/L protease inhibitor cocktail. The intracellular HCV RNA was quantified with a real time one-step reverse-transcription polymerase chain reaction (RT-PCR).
2.2.2. Cytotoxicity assay [15]Huh7.5 cellswere seeded into 96-well plates (Costar) at a density of 3.0 × 104 cells/cm2. After 24 h of incubation, fresh culture mediumcontaining test compounds at various concentrationswere added. 72 h later, cytotoxicity was evaluated with 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT).
2.2.3. Pharmacokinetic studiesAnimals were purchased from the Institute of Laboratory Animal Sciences, Chinese Academy of Medical Sciences, Beijing, China. All experimental procedures were approved by the Biomedical Ethics Committee of the Chinese Academy of Medical Sciences for Animal Use and Protection. Three male SD rats with weights in the range of 180-220 g were used in each study. Each rat was administered orally with compounds at a dose of 25 mg/kg. Blood samples (0.3 mL) were collected at 0, 0.25, 0.5, 1.0, 1.5, 2.0, 4.0, and 6.0 h and were immediately centrifuged to separate the plasma fraction. The plasma samples obtained were stored at S20 ℃ until analysis. Concentration-time profile was obtained for each analyte, and standard noncompartmental analysis was performed on the data using WinNonlin, Version 5.3 to determine the AUC and other noncompartmental parameters.
3. Results and discussionAs depicted in Scheme 1, the matrinic acetic acid 7 was obtained from compound 3 using a four-step sequence including oxidation, esterification, 12-substitution and ester hydrolysis with overall yields of 30%-35%. The desired matrinic ethanamides (8a-b) were prepared using 7 and NH4HCO3 via the mixed anhydride procedures in 70%-80% yields 65%-70%.

|
Download:
|
| Scheme. 1. Synthetic route of matrinic ethanamide derivatives. (a) 10% H2SO4, KMnO4, reflux, 2 h; (b) 2 N MeOH/HCl, reflux, 2 h; (c) RCH2Br, K2CO3, MeCN, r.t., 8 h; (d) 3 mol/L HCl, reflux, 1 h; (e) Boc2O, NH4HCO3, pyridine, MeCN, r.t., 8 h; (f) EDCI, HOBt, TEA, NH2R1, CH2Cl2, r.t. 4 h. | |
As shown in Scheme 2, the matrinic amide products 14a-e were acquired from compound 1 using a five-step sequence including ring-opening, 12-protection using tert-butoxycarbonyl (Boc), amidation of carboxyl, de-protection of Boc and 12-substitution with 30%-40% overall yields. The key intermediates (16a-b) were gained from 1 in ideal yields as described previously 60%-70%, and 18f was produced after the removal of N-Boc of 18d in high yield. The target compounds 23a-b were generated via a five-step procedure, including ester reduction by LiAlH4, selective oxidization of hydroxy group, reductive amination using sodium triacetoxyborohydride 20%-30%. All the final products were purified by flash column chromatography on silica gel with CH2Cl2/ CH3OH as gradient eluents.

|
Download:
|
| Scheme. 2. Synthetic route of matrinic acid/amide and N'-acyl matrinic amine derivatives. (a) 5 mol/L NaOH, reflux, 9 h; (b) Boc2O, MeOH, r.t., 4 h, 10% citric acid, pH 4-5; (c) Boc2O, NH4HCO3, pyridine, MeCN, r.t., 8 h; (d) 2 mol/L HCl/Et2O, 30 min; (e) RCH2Br, K2CO3, MeCN, r.t., 8 h; (f) 2 mol/L MeOH/HCl, reflux, 2 h; (g) RCH2Br, K2CO3, MeCN, r.t., 8 h; (h) 3 mol/L HCl, reflux, 1 h; (i) EDCI, HOBt, TEA, NHR1R2, CH2Cl2, r.t. 8 h; or 2 mol/L HCl/Et2O, 30 min; (j) LiAlH4, THF, r.t., 30 min; (k) DMSO, oxalyl chloride, TEA, CH2Cl2, S78 ℃, 30 min; (l) o-phthalimide, ClCH2CH2Cl, reflux, 4 h, STAB, r.t., overnight; (m) hydrazine hydrate, EtOH; (n) EDCI, HOBt, TEA, NHR1R2, CH2Cl2, r.t. 8 h; or 2 mol/L HCl/ Et2O, 30 min. | |
All the newly synthesized compounds were examined for their anti-HCV activity (EC50) and cytotoxicity (TC50) in human Huh7.5 cells using the specific real-time RT-PCR assay. As an important indicator, the selectivity index (SI) was calculated as a ratio of TC50 to EC50. Anti-HCV ability of each compound was estimated by combining its EC50 and SI values. Nineteen matrinic amide/ ethanamide and acid/amine derivatives and their anti-HCV effects are shown in Table 1.
|
|
Table 1 SAR of matrinic derivatives for anti-HCV activity. |

First, the p-methoxylphenyl on the nitrogen-12 atom was replaced with a -yridyl. The resultant compound 17 exerted an activity with EC50 of 7.54 mmol/L and SI of over 66 (Table 1), better than those of 2. Second, 12-pyridylmethylene and 12-methoxylbenzyl were retained, and SAR investigation was focused on the influence of 4'-carboxyl and shortening length of the 11-side chain, with which a series of new matrinic amide (14a-e) and matrinic ethanamide (8a-b) derivatives were constructed and measured. As described in Table 1, compounds 8a, 14a and 14d-e displayed a reasonable activity with EC50 ranging from 9.6 mmol/L to 97.8 mmol/L and SI from 10 to over 29. These data suggested that replacing carboxyl with amido group might not greatly improve the potency against HCV, regardless of the length of the 11-side chain.
In addition, several substituents were introduced to the N'-end of 8a, and three new N'-substituted matrinic ethanamide (9a-c) analogs were designed and tested. As described in Table 1, all three compounds afforded good potency with EC50 values between 2.12 mmol/L and 5.09 mmol/L, but a high cytotoxicity with CC50 values between 16.3 and 42.7 mmol/L. Then, several groups were also added to the same position of 14d, and a series of N'-substituted matrinic amides 18a-f were also obtained. As anticipated, most of them (18a-d) exhibited promising anti-HCV activity with low micromolar EC50 values from 1.03 mmol/L to 4.89 mmol/L, and better therapeutic windows with SI from 81 to 132. The results indicated that the introduction of a proper substituent at the N'-end of matrinic amides could significantly enhanced the activity against HCV. In addition, two N'-acyl matrinic amine derivatives 23a-b were also designed and tested. As depicted in Table 1, both of them had moderate activity with EC50 values of 14.4 and 5.0 mmol/L, and SI of 8.0 and 22, respectively. The results suggested that introducing a suitable group at the formyl group of matrinic amine derivatives might keep the antiviral activity against HCV.
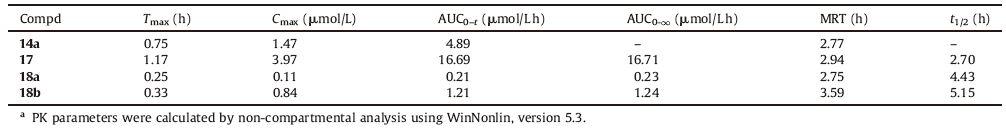
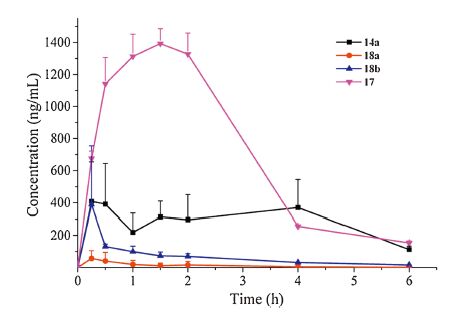
Four representative compounds 14a, 17 and 18a-bwith different structural features were chosen to examine their PK parameters in SD rats at a single dosage of 25mg/kg via oral route. As indicated in Table 2 and Fig. 2, 18a and 18b had a poor PK profile with the area under the curve (AUC) of 0.21 and 1.21 mmol/L h, respectively Similarly, compound 14a demonstrated a moderate PK parameter withAUC of 4.89 mmol/L h. As anticipated, 17 displayed excellentPK properties with the maximum concentration (Cmax) and AUC of 3.97 mmol/L and 16.7 mmol/L h, respectively, indicating a good stability to metabolism in vivo. Compared with 17, compounds 14a and 18a-b displayed an unsatisfying PK profile, which might be resulted from metabolically labile amido group at the N'-end.
|
|
Table 2 PK parametersa of the key compounds in rats after single oral dosing (n = 3) . |
{kind=link}

|
Download:
|
| Figure 2. Mean plasma concentration-versus-time curve of 14a, 17 and 18a–b after oral administration to mice (n = 3) at 25 mg/kg, respectively. | |
{kind=link}
Furthermore, single dose toxicity test for 17 was also carried out in mice. After compound 17 was given orally at a dose of 250, 500 or 1000 mg/kg, respectively, the mice were closely monitored for 7 days. No mouse died in the experiment, indicating that the LD50 value for 17 via oral route was over 1000 mg/kg. In addition, this treatment with 17 showed no effect on body weight of mice as well (data not shown). The results suggested that compound 17 was reasonably safe in vivo.
4. ConclusionTaken together, 19 matrinic amide/ethanamine and matrinic acid/amine derivatives were synthesized and evaluated for their antiviral activity against HCV using compound 2 as the lead with novel mechanism against HCV. The SAR study indicated that the introduction of a suitable group at the N'-end of matrinic amide could greatly improve the anti-HCV potency. Among the newly synthesized compounds, matrinic acid analog 17 demonstrated good potency against HCV with SI of over 66, much better than that of the lead 2. Moreover, it also displayed an excellent PK and safety profile in vivo, indicating good drug-like properties. Thus, compound 17 has been chosen as a novel anti-HCV agent for further investigation with the advantage of decreased chance of causing drug-resistant mutations.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21472246 and 81321004) and the Beijing Natural Science Foundation (No. 7152097) and National Mega-Project for Innovation Drugs (No. 2012ZX09103101-037) .
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.03.006.
| [1] | H.K. Mohd, J. Groeger, A.D. Flaxman, et al. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57 (2013) 1333–1342. DOI:10.1002/hep.26141 |
| [2] | J.H. Fan, J.B. Wang, Y. Jiang, et al. Attributable causes of liver cancer mortality and incidence in China. Asian Pac. J. Cancer Prev. 14 (2013) 7251–7256. DOI:10.7314/APJCP.2013.14.12.7251 |
| [3] | X. Xiang, J. Lu, Z. Dong, et al. Viral sequence evolution in Chinese genotype, 1b chronic hepatitis C patients experiencing unsuccessful interferon treatment. Infect. Genet. Evol. 11 (2011) 382–390. DOI:10.1016/j.meegid.2010.11.011 |
| [4] | S. Susser, C. Welsch, Y. Wang, et al. Characterization of resistance to the protease inhibitor boceprevir in hepatitis C virus-infected patients. Hepatology 50 (2009) 1709–1718. DOI:10.1002/hep.23192 |
| [5] | C. Sarrazin, T.L. Kieffer, D. Bartels, et al. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132 (2007) 1767–1777. DOI:10.1053/j.gastro.2007.02.037 |
| [6] | A.S. Lok, D.F. Gardiner, E. Lawitz, et al., Preliminary study of two antiviral agents for hepatitis C genotype 1, N. Engl. J. Med. 366 (2012) 216-224. |
| [7] | E. Vispo, P. Barreiro, V. Soriano. Pharmacokinetics of new oral hepatitis C antiviral drugs. Expert Opin. Drug Metab. Toxicol. 9 (2013) 5–16. DOI:10.1517/17425255.2013.729577 |
| [8] | S.M. McConachie, S.M. Wilhelm, P.B. Kale-Pradhan. New direct-acting antivirals in hepatitis C therapy: a review of sofosbuvir, ledipasvir, daclatasvir, simeprevir, paritaprevir, ombitasvir and dasabuvir. Expert Rev. Clin. Pharmacol. 8 (2016) 1–16. |
| [9] | J. Guedj, H. Dahari, R.T. Pohl, et al. Understanding silibinin's modes of action against HCV using viral kinetic modeling. J. Hepatol. 56 (2012) 1019–1024. DOI:10.1016/j.jhep.2011.12.012 |
| [10] | A.M. Lam, C. Espiritu, S. Bansal, et al. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob. Agents Chemother. 56 (2012) 3359–3368. DOI:10.1128/AAC.00054-12 |
| [11] | T.T. Jong, M.R. Lee, Y.C. Chiang, S.T. Chiang, Using LC/MS/MS to determine matrine, oxymatrine, ferulic acid, mangiferin, and glycyrrhizin in the Chinese medicinal preparations Shiau-feng-saan and Dang-guei-nian-tong-tang, J. Pharm. Biomed. Anal. 40 (2006) 472-477. |
| [12] | N.N. Du, Z.G. Peng, C.W. Bi, et al., N-substituted benzyl matrinic acid derivatives inhibit hepatitis C virus (HCV) replication through down-regulating host heatstress cognate 70 (Hsc70) expression, PLOS ONE 8 (2013) e58675. |
| [13] | N.N.Du, X. Li, Y.P.Wang, et al., Synthesis, structure-activity relationshipandbiological evaluationofnovelN-substitutedmatrinicacidderivatives ashostheat-stress cognate 70 (Hsc70) down-regulators, Bioorg. Med. Chem. Lett. 21 (2011) 4732-4735. |
| [14] | Z.G. Peng, B. Fan, N.N. Du, et al., Small molecular compounds that inhibit hepatitis C virus replication through destabilizing heat shock cognate 70 messenger RNA, Hepatology 52 (2010) 845-853. |
| [15] | Y.H. Li, Z.G. Peng, L.M. Gao, et al. Synthesis and biological evaluation of sophocarpinic acid derivatives as anti-HCV agents. Acta Pharm. Sin. B 4 (2014) 212–307. |
| [16] | A. Valentina, F. Vilius, K. Matthew, et al., (S)-5-Pyrrolidin-2-yl-1H-tetrazole, Org. Synth. 85 (2008) 72-87. |
| [17] | N. Suzuki, T. Suzuki, Y. Ota, et al., Design, synthesis, and biological activity of boronic acid-based histone deacetylase inhibitors, J. Med. Chem. 52 (2009) 2909-2922. |
| [18] | S. Tang, L. Kong, Y. Li, et al. Novel N-benzenesulfonyl sophocarpinol derivatives as coxsackie B virus inhibitors. ACS Med. Chem. Lett. 6 (2015) 183–186. DOI:10.1021/ml500525s |
| [19] | C. Bi, C. Zhang, Y. Li, et al. Synthesis and biological evaluation of sophoridinol derivatives as a novel family of potential anticancer agents. ACS Med. Chem. Lett. 5 (2014) 1225–1229. DOI:10.1021/ml500289h |