 2016, Vol. 27
2016, Vol. 27 



b Laboratory of Cardiometabolic Molecular Medicine, Institute of Molecular Medicine, Peking University, Beijing 100871, China ;
c College of Life Sciences and Technology, Huazhong University of Science and Technology, Wuhan 430074, China
Glucagon is secreted from pancreatic alpha-cells and enters the circulating blood. After binding to the glucagon receptor, glucagon activates adenylyl cyclase in the hepatocyte plasma membrane and triggers glycogenolysis via a cAMP-related signaling pathway, ensuring glucose homeostasis [1]. In the advanced type 1 diabetes, uncontrolled high glucagon levels are the cause of patients’ death [2]. In type 2 diabetes, insulin- and glucose-induced suppression of glucagon secretion is markedly impaired in patients [3]. Meanwhile, hyperglycemia requires unsuppressible hyperglucagonemia from insulin-resistant alpha-cells in rodent models [4]. Although the bihormonal-abnormality view [5] is still the mainstream view, more and more studies have demonstrated the reasonable properties of the glucagon-centric view in the development of diabetes [6-8]. In the islets of the pancreas, the alpha- and betacells are adjacent to each other. Abundant fenestrated blood capillaries are interspersed among the alpha- and beta-cells [9]. It has been well established that glucagon stimulates insulin secretion in various in vitro and in vivo models [10, 11]. This insulinotropic effect in beta cells occurs through two different pathways. The first pathway is that the endogenous glucagon secreted from the alpha-cells can accumulate in the intercellular space at a certain concentration and act on neighboring beta cells locally. The endogenous glucagon can also enter into the capillaries through the blood circulation, which is defined as exogenous glucagon. The second pathway is that the exogenous glucagon can act on the beta-cells near the capillaries globally. However, an amplifying glucagon signal from neighboring alpha-cells cannot trigger additional glucose-stimulated insulin secretion (GSIS) in the perfused rat pancreas [12]. It is difficult to estimate a single effect of glucagon-induced insulin secretion because the local glucagon effect on glucose-induced insulin release has reached saturation [12]. To clearly understand the mechanism, we designed experiments to switch the stimulation using the following procedure: mouse islets were perfused with an extracellular solution containing 3 μmol/L glucose and then with 20 μmol/L glucose to stimulate insulin secretion, followed by 0 μmol/L glucose to stimulate glucagon secretion. Using this procedure, the effect of glucagon-induced insulin secretion alone can be measured in the 0 μmol/L glucose environment, where there is no GSIS in beta-cells. The time interval measured by a radioimmunoassay is no more than 15 s to detect the precise changes in insulin and glucagon secretion. Here, we developed a quick, small volume, multi-channel PDMS microchip from the Mohammed et al.’s perfusion system [13].
Although glucagon exhibits its insulinotropic actions by activating cAMP through the glucagon or GLP-1 receptors expressed directly on the beta-cell surface [14], the receptor selectivity is still not clear. In this study, we used the PDMS microchip to clarify whether endogenous and exogenous glucagon utilize different receptors to promote insulin secretion, which will provide a better understanding of the glucagon-centric view in diabetes.
2. Experimental 2.1. Microchip assembly and testingA flow diagram of the fabrication of the microchip was depicted in Fig. 1S in Supporting information[1TD$DIF]. Venoclysis needles were used to connect the microchip and the external syringe pumps, and each inlet was controlled by an independent syringe pump. The solution-filled syringes were wrapped in a homemade heating element and heated to 37 ℃. A venoclysis needle was inserted into the outlet of the microchip to collect the fluid. After the cells/islets and extracellular solution were introduced, a polymethyl methacrylate (PMMA) cover was used to seal the perfusion chamber.
To assess the fluid exchange in the chamber, the fluorescence intensity indifferent regions of the perfusion chamber was measured over time. The solutions were perfused from different inlets at a flow rate of 1.5 mL/min. The intensity values of the different regions in the perfusion microchip were measured during the perfusion of fluorescein isothiocyanate (FITC, 2 μmol/L, Sigma, MI, USA) and deionized water (DI) water. Images were captured with 40×, numerical aperture (NA) 0.95 AIR objective of the widefield fluorescent microscope under the same experimental conditions to detect the intensity values. The fluorescent signals were acquired using Andor iXon3 888 EMCCD cameras controlled by the MetaMorph software (Molecular Devices) at a sampling rate of 2 Hz.
2.2. INS-1 cell culture and calcium imagingINS-1 cells (Insulin-producing beta-cell line) were cultured in complete medium composed of RPMI 1640 medium (containing 11.1 μmol/L glucose, Gibco, CA, USA) supplemented with 10% serum (Gibco, CA, USA), 1% sodium pyruvate (Sigma, MI, USA), 0.1% b-mercaptoethanol (Sigma, MI, USA) and a 1% penicillin-streptomycin solution (100 units/mL penicillin, 100 mg/mL streptomycin, Hyclone, UT, USA) at 37 ℃ in a 5% CO2 atmosphere. The INS-1 cells were transfected with pGP-CMV-GCaMP6s (Addgene, MA, USA) using the lipofectamine 2000 reagent (Invitrogen, CA, USA), according to the manufacturer’s instructions. Then, the cells were plated on a poly-L-lysine-coated (Sigma, MI, USA) microchip overnight.
After 24 h, the INS-1 cells were washed in a bath solution containing 136.0 μmol/L NaCl, 4.2 μmol/L KCl, 2.4 μmol/L CaCl2, 1.2 μmol/L KH2PO4, 1.2 μmol/L MgSO4, 4.0 μmol/L glucose, 10.0 μmol/L HEPES and 1.0 μmol/L L-glutamine (pH 7.4). Throughout the calcium imaging experiments, the INS-1 cells were perfused with an extracellular solution containing 3 μmol/L glucose and then 20 μmol/L glucose, followed by 0 μmol/L glucose or 100 nmol/L glucagon (Sigma, MI, USA) to stimulate the calcium response.
We used an Olympus IX81 inverted microscope equipped with a 40×, NA 0.95, air objective (Olympus, Tokyo, Japan). The fluorescent signals were acquired at a sampling rate of 2 Hz. A 473 nm laser was used to excite GCaMP6s.
2.3. Islet isolation and cultureThe handling of the mice and the experimental procedures were conducted according to protocols and guidelines that were approved by the Animal Ethics Committee of Xiamen University (XMULAC20120030). The mice were sacrificed by cervical dislocation. Collagenase P (0.5 mg mL-1 , Roche Diagnostics, IN, USA) was dissolved in Hanks’ buffer and injected into the pancreatic duct after the abdominal cavity was opened. The pancreas was then removed and gently digested for 20 min at 37 ℃ in an incubator. After shaking and washing in collagenase-free Hanks’ solution, the isolated islets were plated on small chambers in RPMI 1640 medium with 11.1 μmol/L glucose, supplemented with 10% fetal bovine serum and a 1% penicillin-streptomycin solution at 37 ℃ in a 5% CO2 atmosphere.
2.4. Perfusion and radioimmunoassay (RIA)To assay insulin and glucagon release, 80 islets from C57BL/6 J mice were pre-incubated in Krebs’ KRBB solution containing 135.0 μmol/L NaCl, 4.7 μmol/L KCl, 10.0 μmol/L HEPES, 3.0 μmol/L glucose, 1.2 μmol/L KH2PO4 and 5.0 μmol/L NaHCO3 for 30 min at 37 ℃. Then, the 80 islets were suck into a microchip under a dissection microscope to avoid damaging the islets. The mouse islets were perfused with an extracellular solution containing 3 μmol/L glucose and then with 20 μmol/L glucose, followed by 0 μmol/L glucose. The solution was collected on ice at different time points. The insulin and glucagon levels were quantified by radioimmunoassay with commercially available kits, according to the manufacturer’s specifications (Human Insulin Radioimmunoassay Kit and Human Glucagon Radioimmunoassay Kit from Beijing North Institution of Biological Technology). Glucagon receptor antagonist II (Millipore, CA, USA) and Exendin (9-39) (Sigma, MI, USA) were also used.
2.5. StatisticsThe results were analyzed using the Igor Pro software (Wavemetrics, Lake Oswego, OR). The averaged results were presented as the means ±SEM from the indicated number of experiments. Statistical significance was evaluated by either Student’s t-test for single Gaussian distributed datasets or the Mann- Whitney rank sum test for non-single Gaussian distributed datasets. The asterisks *, **, and *** denoted statistical significance with P values less than 0.05, 0.01, and 0.001, respectively. All data were from three independent experiments.
3. Results and discussionGlucagon and the glucagon receptors have been studied in different cell types and tissues for many years. The expression cloning and signaling properties of the rat glucagon receptor have improved our understanding of glucagon action. The glucagon receptor is encoded by a 485-amino acid protein, and it Exhibits 42% amino acid identity with the GLP-1 receptor [15]. The glucagon and GLP-1 peptides also Exhibit 47% homology [14, 16]. Structurally, the glucagon receptor core domain contains two or more subsegments that determine glucagon specificity, whereas the GLP-1 receptor core domain also has low affinity for glucagon [14]. In islet beta-cells, the glucagon receptors stimulate insulin secretion by triggering the cAMP pathway. The GLP-1 receptor signaling pathway mediates insulin gene transcription and biosynthesis by activating both PKA-dependent and -independent signaling pathways [17-19]. There is evidence that glucagon enhances insulin secretion in high glucose condition by activating beta-cell signaling via both the glucagon and GLP-1 receptors [12]. However, it remains unclear whether endogenous and exogenous glucagon have different insulinotropic effects, which are mediated by both the glucagon and GLP-1 receptors.
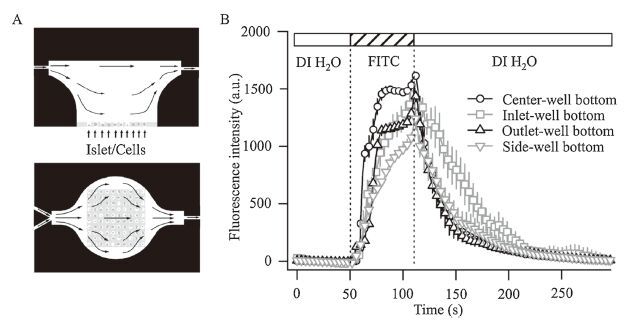
3.1. Characterization of microchipsTo address this question, a PDMS microchip was designed. Fig. 1A shows a longitudinal view and cross-section of the PDMS chamber and channels. The different stimulation solutions were injected into different channels. Only one channel was used each time, and the other channels kept closed. The fluorescent intensity values of the different regions in the perfusion microchip were measured during the perfusion of FITC and DI water. The fluorescent intensity variations were clearly explained by the complete exchange of the stimulus solution. The fluorescent intensity values at the center-well bottom peaked at approximately 25 s, and 75% of the FITC solution was replaced in approximately 30 s (Fig. 1B). Therefore, the fluid flow within the perfusion microchip was fast enough to switch from one solution to the next solution, which ensured that the islets or cells were able to sense a homogeneous stimulus. It is important to characterize the microfluidic device to reflect an accurate secretion plot [13]. Compared with the microfluidic device used for islets research in Mohammed et al.’s study [13], we optimized the corner that had been ignored but significantly influenced the liquid in both the channel and the reaction chamber. Further improvements to the fabrication procedure not only reduced the difficulty of the production process but also heightened the integrity of the microchip.

|
Download:
|
| Figure 1. (A) a longitudinal view and cross-section of the PDMS chamber and channels. The different stimulation solutions were injected into different channels. Only one channel was used each time, and the other channels kept closed. (B) The fluorescence intensity values in different regions of the perfusion microchip were measured during the perfusion of FITC and DI water. The complete exchange of its stimulus solution was clearly explained by the fluorescence intensity variations in the figure. The intensity values at the center-well bottom peaked at approximately 25 s, and 75% of the FITC solution was replaced in approximately 30 s. Therefore, the fluid flowwithin the perfusion microchip was fast enough to switch from one solution to the next solution, which ensured that the islets or cells were able to sense a homogeneous stimulus. It is important to characterize the microfluidic device to reflect an accurate secretory plot. n = 4 for each group. | |
{kind=link}
3.2. Higher calcium response induced by glucagon in INS-1 cells
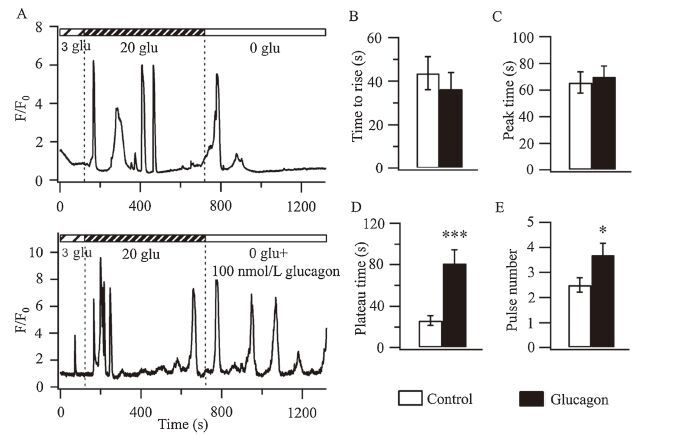
Next, we cultured the INS-1 cells in the perfusion chamber. When stimulated with high glucose, the INS-1 cells were depolarized, which triggered calcium oscillations and pulsatile insulin release. We used the glucose-stimulated calcium transient as an indicator to detect the cells’ function. We tested the changes in the transients from 3 μmol/L to 20 μmol/L and then 0 μmol/L glucose, because Hope et al.’s study demonstrated that an insulin switch-off signal of either endogenous or exogenous insulin signaled the alpha-cells to respond to the 0 μmol/L glucose conditions [20]. In beta-cells, cAMP augmented the glucoseinduced cytosolic calcium oscillations and insulin secretion by acting through PKA-dependent pathways [21, 22]. The treatment with 100 nmol/L glucagon induced more calcium oscillations (Fig. 2A). Then, we normalized the intensity of the calcium signals over the basal signal that was present within the 3 μmol/L glucose perfusion. We determined the maximal amplitude of the fluorescent intensity of calcium during 0 μmol/L glucose stimulation and established it as the peak amplitude. We designated the first time point at which the increase in the fluorescent intensity of calcium was greater than 50% of the maximal amplitude as the time to rise, which defined the relative speed of the calcium response. We also designated the time point when the increase in the fluorescent intensity of calcium reached maximal amplitude as the peak time, which defined the maximal amplitude of the calcium response. Additionally, we defined the time points that were greater than 50% of the maximal amplitude of the calcium fluorescence as the plateau time to describe how long the calcium levels remained elevated. There was no significance difference in either the time to rise or peak time (Fig. 2B and C). However, the plateau time (Fig. 2D) and pulse number (Fig. 2E) were markedly increased upon perfusion of 100 nmol/L glucagon, which suggested that the cells’ function was enhanced upon perfusion of exogenous glucagon.

|
Download:
|
| Figure 2. Glucagon enhances the calcium response in INS-1 cells after switching to a 0 μmol/L glucose solution. (A) Calcium was measured at the single INS-1 cell. The time interval is 500 ms. Each trace is representative of eight recordings from similar experiments. F is the measured fluorescence intensity and F0 is the baseline fluorescence intensity. The average time to rise (B), peak time (C), plateau time (D) and pulse number (E) during the perfusion of a 0 μmol/L glucose solution or a 0 μmol/L glucose solution with 100 nmol/L glucagon. n = 8 for each group. | |
{kind=link}
3.3. Glucagon secretion precedes insulin on 0 μmol/L glucose
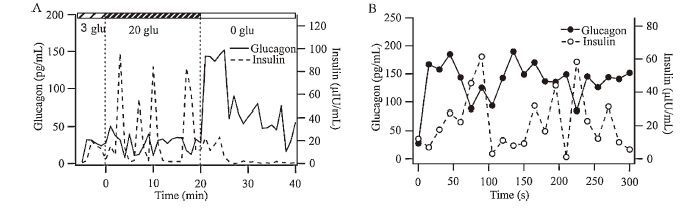
Moreover, we investigated whether glucagon exhibited the same stimulatory effect on the islets. During perfusion with 3 μmol/L glucose, insulin and glucagon release were relatively stable, with a few detectable pulses when samples were taken at 1 min intervals (Fig. 3A). The introduction of 20 μmol/L glucose resulted in larger pulses of insulin and glucagon release. The first rapid glucagon pulse was ahead of the early phase of the insulin pulse. Then, insulin and glucagon release alternated. After switching to 0 μmol/L glucose, glucagon release was significantly increased, while insulin release was inhibited. To observe the precise temporal relationship, samples were taken at 15 s intervals over the first 300 s during the 0 μmol/L glucose perfusion (Fig. 3B), compared with 1 min in other studies [20, 23]. Thus, the real-time glucagon and insulin concentrations can be detected more accurately, which greatly reduces the possibility of artifacts. Then we found that the insulin response was delayed by 63 ± 6.27 s compared with the glucagon response, which suggested the insulinotropic effect of glucagon in the 0 μmol/L glucose condition.

|
Download:
|
| Figure 3. Glucagon secretion precedes insulin secretion in a 0 μmol/L glucose solution. (A) Insulin and glucagon were both measured from the 80 perfused mice islets. The time interval is 1 min. This trace is representative of five recordings from similar experiments. (B) Enlarged time scale of the first 300 s during insulin and glucagon secretion in solutions from 20 to 0 μmol/L glucose. The time interval is 15 s. This trace is representative of five recordings. Glucagon, solid line, black filled circle. Insulin, dashed line, open circle. | |
{kind=link}
3.4. Different insulinotropic effect by endogenous and exogenous
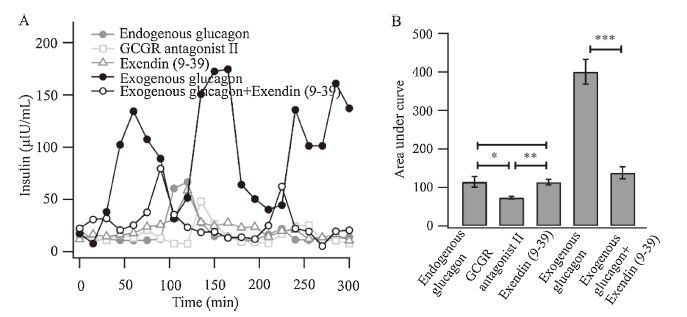
glucagon We continued the switching procedure at 15 s intervals and focused on the initial 300 s during the 0 μmol/L glucose perfusion. Insulin release was precisely measured using different agents (Fig. 4A). 1 μmol/L glucagon receptor (GcgR) Antagonist II (a GcgRspecific antagonist) significantly inhibited the endogenous glucagon- induced insulin release (Fig. 4B). However, a GLP-1 receptorspecific antagonist, Exendin (9-39) (1 μmol/L), had no effect on insulin release (Fig. 4B). These results indicated that endogenous glucagon promotes insulin release from the beta cells through the GcgR pathway. Although endogenous glucagon could enhance insulin release from beta-cells in mouse islets, a higher concentration of 100 nmol/L exogenous glucagon still had the ability to further enhance insulin release, which could be completely inhibited by Exendin (9-39) (Fig. 4B). This indicated that the additional insulin secretion was trigged via the GLP-1 receptor pathway and was three-fold higher than the insulin secretion via the glucagon receptor pathway. Collectively, these results demonstrate that endogenous and exogenous glucagon utilize different receptors to promote insulin secretion in mice.

|
Download:
|
| Figure 4. Endogenous and exogenous glucagon utilize different receptors to promote insulin secretion in mice. (A) The insulin secretion in 20-0 μmol/L glucose was precisely measured for 300 s following the different treatments. This trace is representative off our recordings from similar experiments. Endogenous glucagon, filled circle, dark gray. GcgR antagonist II, open square, light gray. Exendin (9-39), open triangle, dark gray. Exogenous glucagon, filled circle, black. Exogenous glucagon and Exendin (9-39), open circle, black. (B) Area integral of the insulin secretion in response to the various agents. n = 4 for each group. | |
{kind=link}
By considering the case of diabetes, the number of hepatic glucagon receptors in diabetic db/db mice was significantly higher than in the normal db/+ mice [24]. Similarly, in rat islets, the expression of the glucagon receptor mRNA was up-regulated in response to 20μmol/L glucose [25]. However, the expression of the GLP-1 receptor mRNA in rat islets was down-regulated [25, 26]. Under some extreme conditions, such as hyperglucagonemia in glucagonoma syndrome [27], the insulinotropic effect of endogenous glucagon on insulin secretion is saturated, as a protective mechanism, and excess exogenous glucagon can promote insulin secretion via GLP-1 receptor pathway to maintain glucose homeostasis. In diabetes, the high glucose-induced increase in the expression of the glucagon receptor and the decrease in the expression of the GLP-1 receptor might suggest that the beta-cells’ sensitivity to glucagon was altered. It is possible that diabetic individuals have lost the differential receptor selectivity to glucagon
4. ConclusionsIn summary, we have developed a fast, precise, switchable PDMS microchip. Using this microchip, we confirmed that glucagon could enhance the calcium response of the beta cells in the 0 μmol/L glucose condition. We also found that endogenous and exogenous glucagon utilize different receptors to promote insulin secretion in mice as a protective mechanism. The microchip can be further used to explore the mechanism in diabetes. Thus, the application of glucagon receptor antagonists and GLP-1 analogs carefully considered in future diabetes therapy.
AcknowledgmentThis work was supported by the National Natural Science Foundation of China (Nos. 41076064, 31371444).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.05.023.
| [1] | E.W. Sutherland, C. De Duve. Origin and distribution of the hyperglycemicglycogenolytic factor of the pancreas. J. Biol. Chem. 175 (1948) 663–674. |
| [2] | R.H. Unger, A.D. Cherrington. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J. Clin. Invest. 122 (2012) 4–12. DOI:10.1172/JCI60016 |
| [3] | B.E. Dunning, J.E. Gerich. The role of a-cell dysregulation in fasting and postprandial hyperglycemia in type, 2 diabetes and therapeutic implications. Endocr. Rev. 28 (2007) 253–283. DOI:10.1210/er.2006-0026 |
| [4] | Y. Lee, E.D. Berglund, X.X. Yu, et al. Hyperglycemia in rodent models of type, 2 diabetes requires insulin-resistant alpha cells. Proc. Natl. Acad. Sci. U. S. A. 111 (2014) 13217–13222. DOI:10.1073/pnas.1409638111 |
| [5] | R.H. Unger, L. Orci. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet 305 (1975) 14–16. DOI:10.1016/S0140-6736(75)92375-2 |
| [6] | A.D. Baron, L. Schaeffer, P. Shragg, O.G. Kolterman. Role of hyperglucagonemia in maintenance of increased rates of hepatic glucose output in type II diabetics. Diabetes 36 (1987) 274–283. DOI:10.2337/diab.36.3.274 |
| [7] | H.Y. Gaisano, P.E. Macdonald, M. Vranic. Glucagon secretion and signaling in the development of diabetes. Front. Physiol. 3 (2012) 349. |
| [8] | S. Malmgren, B. Ahré n. Evidence for time dependent variation of glucagon secretion in mice. Peptides 76 (2016) 102–107. DOI:10.1016/j.peptides.2016.01.008 |
| [9] | L.R. Nyman, K.S. Wells, W.S. Head, et al. Real-time, multidimensional in vivo imaging used to investigate blood flow in mouse pancreatic islets. J. Clin. Invest. 118 (2008) 3790–3797. DOI:10.1172/JCI36209 |
| [10] | E. Samols, G. Marri, V. Marks. Interrelationship of glucagon, insulin and glucose: the insulinogenic effect of glucagon. Diabetes 15 (1966) 855–866. DOI:10.2337/diab.15.12.855 |
| [11] | P. Huypens, Z. Ling, D. Pipeleers, F. Schuit. Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia 43 (2000) 1012–1019. DOI:10.1007/s001250051484 |
| [12] | K. Moens, D. Flamez, C. Van Schravendijk, et al. Dual glucagon recognition by pancreatic β-cells via glucagon and glucagon-like peptide, 1 receptors. Diabetes 47 (1998) 66–72. DOI:10.2337/diab.47.1.66 |
| [13] | J.S. Mohammed, Y. Wang, T.A. Harvat, J. Oberholzer, D.T. Eddington. Microfluidic device for multimodal characterization of pancreatic islets. Lab Chip 9 (2009) 97–106. DOI:10.1039/B809590F |
| [14] | S. Runge, B.S. Wulff, K. Madsen, H. Bräuner-Osborne, L.B. Knudsen. Different domains of the glucagon and glucagon-like peptide-1 receptors provide the critical determinants of ligand selectivity. Br. J. Pharmacol. 138 (2003) 787–794. DOI:10.1038/sj.bjp.0705120 |
| [15] | L.J. Jelinek, S. Lok, G.B. Rosenberg, et al. Expression cloning and signaling properties of the rat glucagon receptor. Science 259 (1993) 1614–1616. DOI:10.1126/science.8384375 |
| [16] | L.B. Knudsen, D. Kiel, M. Teng, et al. Small-molecule agonists for the glucagon-like peptide, 1 receptor. Proc. Natl. Acad. Sci. U. S. A. 104 (2007) 937–942. DOI:10.1073/pnas.0605701104 |
| [17] | D.J. Drucker, J. Philippe, S. Mojsov, W.L. Chick, J.F. Habener. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc. Natl. Acad. Sci. U. S. A. 84 (1987) 3434–3438. DOI:10.1073/pnas.84.10.3434 |
| [18] | B. Portha, C. Tourrel-Cuzin, J. Movassat. Activation of the GLP-1 receptor signalling pathway: a relevant strategy to repair a deficient beta-cell mass. Exp. Diabetes Res. 2011 (2011) 376509. |
| [19] | P.E. MacDonald, W. El-Kholy, M.J. Riedel, et al. The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes 51 (2002) S434–S442. DOI:10.2337/diabetes.51.2007.S434 |
| [20] | K.M. Hope, P.O.T. Tran, H.R. Zhou, et al. Regulation of α-cell function by the β-cell in isolated human and rat islets deprived of glucose: the “switch-off” hypothesis. Diabetes 53 (2004) 1488–1495. DOI:10.2337/diabetes.53.6.1488 |
| [21] | G. Tian, S. Sandler, E. Gylfe, A. Tengholm. Glucose- and hormone-induced cAMP oscillations in α- and β-cells within intact pancreatic islets. Diabetes 60 (2011) 1535–1543. DOI:10.2337/db10-1087 |
| [22] | L.R. Landa Jr., M. Harbeck, K. Kaihara, et al. Interplay of Ca2+ and cAMP signaling in the insulin-secreting MIN6 b-cell line. J. Biol. Chem. 280 (2005) 31294–31302. DOI:10.1074/jbc.M505657200 |
| [23] | B. Hellman, A. Salehi, E. Grapengiesser, E. Gylfe. Isolated mouse islets respond to glucose with an initial peak of glucagon release followed by pulses of insulin and somatostatin in antisynchrony with glucagon. Biochem. Biophys. Res. Commun. 417 (2012) 1219–1223. DOI:10.1016/j.bbrc.2011.12.113 |
| [24] | H. Yoowarren, A.G. Willse, N. Hancock, et al. Regulation of rat glucagon receptor expression. Biochem. Biophys. Res. Commun. 205 (1994) 347–353. DOI:10.1006/bbrc.1994.2671 |
| [25] | N. Abrahamsen, E. Nishimura. Regulation of glucagon and glucagon-like peptide-1 receptor messenger ribonucleic acid expression in cultured rat pancreatic islets by glucose, cyclic adenosine, 30, 50-monophosphate, and glucocorticoids. Endocrinology 136 (1995) 1572–1578. |
| [26] | G. Xu, H. Kaneto, D.R. Laybutt, et al. Downregulation of GLP-1 and GIP receptor expression by hyperglycemia: possible contribution to impaired incretin effects in diabetes. Diabetes 56 (2007) 1551–1558. DOI:10.2337/db06-1033 |
| [27] | A.P. van Beek, E.R. de Haas, W.A. van Vloten, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur. J. Endocrinol. 151 (2004) 531–537. DOI:10.1530/eje.0.1510531 |