 2016, Vol. 27
2016, Vol. 27 



b Xintai Institute of Skin Diseases, Xintai 271200, China ;
c The Second Hospital of Shandong University, Jinan 250033, China ;
d Faculty of Pharmaceutical Sciences, School of Health Sciences, University of Iceland, Iceland
Multidrug resistance (MDR) has long been recognized as a serious problem in cancer treatment. One of the major mechanisms of MDR in tumor cells is the over-expression of P-glycoprotein (P-gp) [1]. Therefore, there is great clinical interest in developing compounds that overcome resistances with lower host toxicity [2]. Several types of compounds have been identified as P-gp modulators, but most of them were reported to show toxicity at the pharmacological dose required to achieve significant MDR reversal [3, 4]. Thus, P-gp inhibitors of plant origin have the potential to be developed as MDR reversing agents.
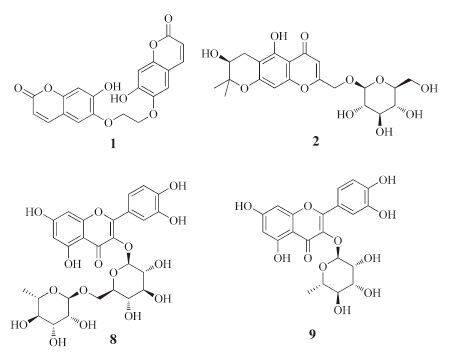
In a previous study, we reported that a series of coumarins from the methanol extract of Cicuta virosa L. possess potent multidrug resistance reversing activity [5]. In connection with our interest in this plant, two new compounds were isolated and identified, together with seven known compounds (Fig. 1). In addition, the chemical constituents were evaluated for their multidrug resistance reversing activity on human myelogenous leukemia cells.
2. Experimental 2.1. GeneralUV spectra were recorded in MeOH on a Agilent 8453E UV-vis spectroscopy system. The IR spectra were measured in KBr on a Thermo Nicolet NEXUS 470 FT-IR spectrometer. NMR spectra were recorded on a Bruker AV 600 MHz NMR spectrometer with TMS as an internal standard. HRESIMS data were obtained from - Fisher Scientific LTQ-Orbitrap XL. HPLC was performed on Agilent 1260 HPLC system. Optical rotation data were measured by a GYROMAT-HP polarimeter. The silica gel GF254 used for TLC was supplied by the Qingdao Marine Chemical Co., Ltd., Qingdao, China. Sephadex LH-20 and silica gel (200-300 mesh) used for column chromatography were supplied by Pharmacia Biotech AB, Uppsala, Sweden and Qingdao Marine Chemical Co., Ltd., Qingdao, China, respectively. Spots of TLC were visualized within I2 vapor or by spraying with H2SO4/EtOH 1:9 followed by heating. All solvent ratios are measured v/v.

|
Download:
|
| Figure 1. Structures of compounds [2TD$DIF]1, 2, 8, and 9 isolated from C. virosa. | |
2.2. Plant material
C. virosa Linnaeus was collected in July 2012 in Heilongjiang Province, China, and was identified by Prof. Xueshen Wen, School of Pharmaceutical Sciences, Shandong University. A voucher specimen (No. 201207CV) has been deposited at the Laboratory of Natural Products Chemistry, School of Pharmaceutical Sciences, Shandong University, China.
2.3. Extraction and isolationThe air-dried powder of the plant material of C. virosa L. (3.0 kg) was extracted with EtOH/H2O (3 × 3 L, 95:5, v/v) under condition of reflux. The combined EtOH extracts were concentrated in vacuum to yield the crude material (780 g), which was then successively partitioned with petroleum ether (PE; 210 g), AcOEt (140 g), and BuOH (240 g). The AcOEt fraction was chromatographed over silica gel, eluted with a solvent gradient system (PE/ acetone 5:1-1:1) to produce fractions A-D. Fraction A (12 g) was subjected to column chromatography on silica gel eluted by PE/ acetone (50:1-10:1), to give three sub-fractions (A1-A3). Subfraction A2 (220 mg) was separated by semi-preparative HPLC (MeOH/H2O 80:20, 1.5 mL/min, ) to give 4 (38.1 mg, tR 21.5 min) and 5 (60.5 mg, tR 24.2 min). Fraction C (345 mg) was further separated via column chromatography on silica gel with PE/ acetone (30:1-10:1) to yield five sub-fractions (C1-C5). Subfraction C3 (35 mg) was purified by Sephadex LH-20, using CH3Cl/ CH3OH (1:1) to yield compound 1 (25 mg). Fraction D (23 g) was further separated on column chromatography on silica gel with PE/ acetone (10:1-5:1) to yield six sub-fractions (D1-D6). Sub-fraction D2 (80.4 mg) was purified by semi-preparative HPLC (MeOH/H2O 70:30, 1.5 mL/min) to give 3 (46.3 mg, tR 18.5 min). The BuOH extract was applied to column chromatography on silica gel eluted by CH2Cl2/CH3OH (200:1-1:1) in gradient to give five fractions (E- I). Fraction E (18 g) was subjected to column chromatography on silica gel using CH2Cl2/CH3OH (50:1-10:1), to yield mixtures (E1- E4). Sub-fractions E1 (150.0 mg) and E2 (95.7 mg) were purified by semi-preparative HPLC (CH3OH/H2O 65:35, 1.5 mL/min) to give 2 (35.2 mg, tR 15.4 min) and 6 (43.1 mg, tR 17.3 min), respectively. Fraction G (12.5 g) was applied to column chromatography on silica gel, with CH2Cl2/CH3OH (20:1) as eluent to give two fractions (G1 and G2). Sub-fraction G2 (115 mg) was separated on a Sephadex LH-20 column with CH2Cl2/CH3OH (1:1), to yield compound 8 (35.3 mg). Fraction H (520 mg) was further chromatographed on Sephadex LH-20 (CHCl3/MeOH 1:1) and then purified by semi-preparative HPLC eluted with (MeOH/H2O 50:50, 1.5 mL/ min) to obtain 7 (55.8 mg, tR 12.4 min) and 9 (78.3 mg, tR 14.7 min).
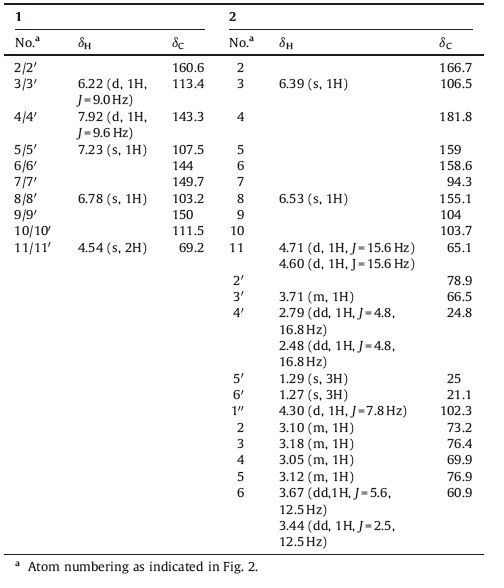
11, 110-Dimer of scopoletin (1): Amorphous colorless powder. UV (MeOH): 230 (3.92), 254 (3.34). IR (KBr, cm-1): 3650, 1710, 1620, 1511. 1H NMR (CDCl3, 600 MHz) and 13C NMR (CDCl3, 150 MHz): see Table 1. HRESIMS: 383.0765 ([M + H]+; C20H15O8+ , calcd. 383.0767).
11-O-β-Glucopyranosyl-hamaudol (2): Amorphous colorless powder. [α]D20 -63 (c 0.11, MeOH). CD (MeOH): Δ ε254 + 5.2, Δ ε232 - 4.2. UV (MeOH): 230 (4.11), 252 (5.05). IR (KBr): 3499, 1655, 1580. 1H NMR (CDCl3, 600 MHz) and 13C NMR (CDCl3, 150 MHz): see Table 1. HRESIMS: 455.1547 ([M + H]+; C21H27O11þ, calcd. 455.1553).
2.4. Sugar identification.D-Glucose (60 mg) and L-cysteine methyl ester hydrochloride (75 mg) were dissolved in pyridine (4 mL) and stirred at 60 ℃ for 1.5 h, and then o-tolyl isothiocyanate (330 μL) was added to the mixture and heated at 60 ℃ for 1.5 h. Separation by HPLC on an C- 18 column eluted with H2O (containing 0.2% TFA)-CH3CN (70:30) gave the derivative S-1. The HRESIMS of S-1 gave a quasi-molecular ion [M + H]+ peak at m/z 447.1246 (calcd. for C18H27N2uO7S2, 447.1254), which was well consistent with our previous experiment [6]. compound 2 was hydrolyzed with HCl and extracted with CH2Cl2. The aqueous layer was passed through a Sephadex LH-20 column and the eluate was concentrated. The residue was dissolved in pyridine (0.4 mL) and stirred with L-cysteine methyl ester (10 mg) for 1.5 h at 60 ℃, and then o-tolyl isothiocyanate (40 μL) was added to the mixture and heated at 60 ℃ for 1.5 h. The reaction mixture was analyzed by HPLC and detected at 250 nm. Analytical HPLC was performed on a Phenomenex C18 column (4.6 × 250 mm) at 25 ℃ using a gradient of CH3CN:0.2%TFA in H2O: 0-25 min (32:68), 25-35 min (from 32:68 to 70:30), and 35- 60 min (70:30) as the mobile phase. Peaks were detected with an Agilent DAD detector. D-Glucose was identified as the sugar moiety of 2 according to the same retention time (tR 14.2 min).
|
|
Table 1 1H NMR and 13C NMR data of 1 and 2 (600MHz for 1H NMR and 150MHz for 13C NMR, in CDCl3, d in ppm, J in Hz). |
{kind=link}
2.5. Biological assays
The human myelogenous leukemia cell line K562, and its multidrug-resistant counterpart K562/A02 were obtained from the Department of Pharmacology, the Institute of Hematology of the Chinese Academy of Medical Sciences (Tianjin, China). K562/A02 cells were maintained in a complete RPMI-1640 medium containing 1 mg/mL doxorubicin (DOX) (Pfizer Italia S.r.l.) at 378 in a humidified atmosphere of 5% CO2. The cells were cultured for two weeks in drug-free medium prior to their use in the experiments. Cells were harvested and seeded into 96-well plates at 2 × 104 cells/well. For cytotoxicity experiments, different concentrations of compounds 1-9 were added into designated wells, and for MDR reversal experiments, different concentrations of DOX were added into designated wells with or without compounds 1-9 (10 mmol/ L). After 48 h, 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) solution was added to each well, and the plate was further incubated for 4 h. The medium was discarded, and 100 mL of DMSO was added into each well to dissolve the formazan crystal. The absorbance in individual wells was determined at 570 nm. IC50 values for compounds 1-9 and doxorubicin (concentration resulting in 50% inhibition of cell growth) were calculated from plotted results using untreated cells as 100%. The reversal fold (RF) values, as potency of reversal, were obtained from fitting the data to RF = IC50 of cytotoxic DOX alone/IC50 of cytotoxic DOX in the presence of the tested compounds [7].
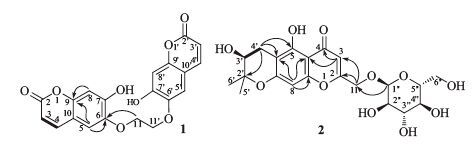
3. Results and discussioncompound 1 was obtained as colorless, amorphous powder. The positive-ion-mode HRESIMS exhibited a quasi-molecular-ion peak at m/z 383.0765 ([M + H]+; calcd. 383.0767) corresponding to the molecular formula C20H14O8. The NMR spectra of 1 indicated a dimeric structure with a total of 15 C-atoms signals observed in the 13C NMR spectra (Table 1). The spectral data of 1 was very similar to those of scopoletin with some notable differences [8, 9]. The 13C NMR spectra of 1 closely resembled that of scopoletin, except for the signal assignable to C-11. The C-11 signal at δC 69.2 showed a downfield shift by 12.8 ppm. The downfield shift of the signal assignable to H-11 was also observed in the 1H NMR spectra. The H-11 signal at δH 4.54 (s, 2H, H-11/110) showed a downfield shift by 0.72 ppm. All of the above data indicated that 1 might be the 11, 110-dimer of scopoletin. It was also supported by the NMR data of another coumarin dimer, diumancal, which was very similar to 1 [10]. Therefore, compound 1 was elucidated as 11, 110-dimer of scopoletin (Fig. 2).

|
Download:
|
| Figure 2. The selected 1H, 1H-COSY ([TD$INLINE] ) and HMBCs (H!C) correlations of 1 and 2. | |
{kind=link}
compound 2 was obtained as a colorless amorphous powder. The HRESIMS exhibited the [M + H]+ peak at m/z 455.1547 (calcd. 455.1553), corresponding to the molecular formula C21H26O11. The 13C NMR spectra exhibited six signals due to a b-glucopyranosyl moiety, and 15 signals attributable to the aglycone moiety, which was similar to those of hamaudol (Table 1) [11]. The signals at δH 6.53 (s, 1H, H-8) and 6.39 (s, 1H, H-3) were characteristic for the chromone of 2. The signals at δH 2.79 (dd, 1H, J = 4.8, 16.8 Hz, H-40a) and δH 2.48 (dd, 1H, J = 4.8, 16.8 Hz, H-40b), 3.71 (m, 1H, H-30), and 1.29, 1.27 (s, each 3H, H-50, 60) can be assigned to a trisubstituted pyran ring. In addition, the signal of anomeric H-atoms appeared at δH 4.30 (d, 1H, J = 7.8 Hz, H-100) with coupling constants characteristic of a b-configuration. After sugar analysis, the presence of a D-glucose was confirmed [6]. The glucose residue in 2 was found to be linked to C-11 since the signal of anomeric proton at δH 4.30 showed a 1H-13C long range correlation with a signal of the C-11 at δC 65.1, in the HMBC spectrum (Fig. 2). The absolute configuration at C-30 of 2 was confirmed as S by comparison of the circular dichrosim (CD) curve with those of hamaudol [11]. The basis of the evidence obtained, compound 2 was assigned to 11-O-β-glucopyranosyl-hamaudol.
The seven known compounds were isobyakangelicin (3) [12], psoralene (4) [13], angelicin (5) [14], prim-O-glucosylangelicain (6) [15], apiosylskimmin (7) [16], rutin (8) [17], and quercetin-3-Ob- D-rhamnoside (9) [18], respectively.
The in vitro cytotoxic activities of the isolated compounds were evaluated against K562 and K562/A02. Compounds 1-9 possessed very weak cytotoxic activities (Table 2). TheMDRreversal effects of compounds 1-9 were further investigated by using the revised 3- (4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-2H-tetrazolium bromide (MTT) method as described in the Experimental Part. Compounds 1, 8, and 9 were most active and exhibited significant MDR reversal effects on K562/A02 cell line, using verapamil as a reference compound.
In conclusion, nine compounds, including two new compounds (1 and 2), were isolated from the C. virosa. The present study about the isolation and identification of nine compounds shows the diversity of chemical constituents in C. virosa. In addition, some of these compounds showed remarkable MDR reversing effects.
AcknowledgmentsFinancial supported from the National Natural Science Foundation of China (Nos. 81202422, 21472113, and 31500280) and the Natural Science Foundation of Shandong Province (No. ZR2012HQ024) are acknowledged.
| [1] | H.J. Jung, S.Y. Chung, J.W. Nam, et al. Inhibition of P-glycoprotein-induced multidrug resistance by a clerodane-type diterpenoid from Sindora sumatrana. Chem. Biodivers. 7 (2010) 2095–2101. DOI:10.1002/cbdv.201000010 |
| [2] | E. Caballero, J.I. Manzano, P. Puebla, et al. Oxazolo[3, 2-α]pyridine. A new structural scaffold for the reversal of multi-drug resistance in Leishmania. Bioorg. Med. Chem. Lett. 22 (2012) 6272–6275. DOI:10.1016/j.bmcl.2012.07.100 |
| [3] | E. Teodori, S. Dei, C. Martelli, S. Scapecchi, F. Gualtieri. The functions and structure of ABC transporters: implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance (MDR). Curr. Drug. Targets 7 (2006) 893–909. DOI:10.2174/138945006777709520 |
| [4] | G.A. Fisher, B.L. Lum, J. Hausdorff, B.I. Sikic. Pharmacological considerations in the modulation of multidrug resistance. Eur. J. Cancer 32a (1996) 1082–1088. |
| [5] | S.Q. Wang, X. Li, X.N. Wang, N.N. Wei, H.X. Lou. Coumarins from Cicuta virosa and their modulating effects on multidrug-resistant (MDR) tumors. Phytochem. Lett. 4 (2011) 97–100. DOI:10.1016/j.phytol.2010.11.007 |
| [6] | S.Q. Wang, D.M. Ren, F. Xiang, et al. Dracotanosides A-D, spermidine glycosides from Dracocephalum tanguticum: structure and amide rotational barrier. J. Nat. Prod. 72 (2009) 1006–1010. DOI:10.1021/np900140s |
| [7] | X. Li, B. Sun, C.J. Zhu, et al. Reversal of p-glycoprotein-mediated multidrug resistance by macrocyclic bisbibenzyl derivatives in adriamycin-resistant human myelogenous leukemia (K562/A02) cells. Toxicol. In Vitro 23 (2009) 29–36. DOI:10.1016/j.tiv.2008.09.015 |
| [8] | T.K. Razdan, S. Harkar, B. Qadri, M.A. Qurishi, M.A. Khuroo. Lupene derivatives from Skimmia laureola. Phytochemistry 27 (1988) 1890–1892. DOI:10.1016/0031-9422(88)80473-4 |
| [9] | T. Iossifova, B. Vogler, I. Kostova. Escuside, a new coumarin-secoiridoid from Fraxinus ornus bark. Fitoterapia 73 (2002) 386–389. DOI:10.1016/S0367-326X(02)00132-6 |
| [10] | A.Z. Abyshev, I.K. Zhurkovich, E.M. Agaev, A.A. Abdulla-zade, A.B. Guseinov. Methods of quality standardization for diumancal parent substance and related medicinal forms. Pharm. Chem. J. 41 (2007) 50–53. DOI:10.1007/s11094-007-0011-8 |
| [11] | H. Sasaki, H. Taguchi, T. Endo, I. Yosioka. The constituents of Ledebouriella seseloides WOLFF. I. Structures of three new chromones. Chem. Pharm. Bull. 30 (1982) 3555–3562. DOI:10.1248/cpb.30.3555 |
| [12] | R.T. Li, A.H. Zhao, Y.H. Sheng, Z. Na, H.D. Sun. Chemical constituents from Schisandra plena. J. Asian Nat. Prod. Res. 7 (2005) 847–852. DOI:10.1080/1028602042000204045 |
| [13] | S.B. Wu, Y. Zhao, H. Fan, et al. New guaiane sesquiterpenes and furanocoumarins from Notopterygium incisum. Planta Med. 74 (2008) 1812–1817. DOI:10.1055/s-0028-1088326 |
| [14] | G. Kavli, K. Midelfart, J. Raa, G. Volden. Phototoxicity from furocoumarins (psoralens) of Heracleum laciniatum in a patient with vitiligo. Action spectrum studies on bergapten, pimpinellin, angelicin and sphondin. Contact Derm. 9 (1983) 364–366. DOI:10.1111/cod.1983.9.issue-5 |
| [15] | B. Zhao, X. Yang, X. Yang, L. Zhang. Chemical constituents of roots of Saposhnikovia divaricata. Chin. J. Chin. Mater. Med. 35 (2010) 1569–1572. |
| [16] | J. Shi, J. Yang, C. Li, D. Zhang. Chemical constituents from Hydrangea paniculata. Chin. J. Chin. Mater. Med. 35 (2010) 3007–3009. |
| [17] | G.Y. Zhail, W.T. Qu, Z.T. Yan, et al. Synthesis, spectral and antioxidant properties of Tin(II)-rutin complex. Chem. Nat. Compd. 50 (2014) 624–628. DOI:10.1007/s10600-014-1039-0 |
| [18] | N.H.T. Le, K.E. Malterud, D. Diallo, et al. Bioactive polyphenols in Ximenia americana and the traditional use among Malian healers. J. Ethnopharmacol. 139 (2012) 858–862. DOI:10.1016/j.jep.2011.12.031 |