 2016, Vol. 27
2016, Vol. 27 



b Department of Chemistry, University of Pennsylvania, Philadelphia 19104-6323, USA ;
c Unité Molécules de Communication et Adaptation des Micro-organismes, Sorbonne Universités, Muséum National d'Histoire Naturelle, Centre National de la Recherche Scientifique (CNRS UMR 7245), Paris 75005, France
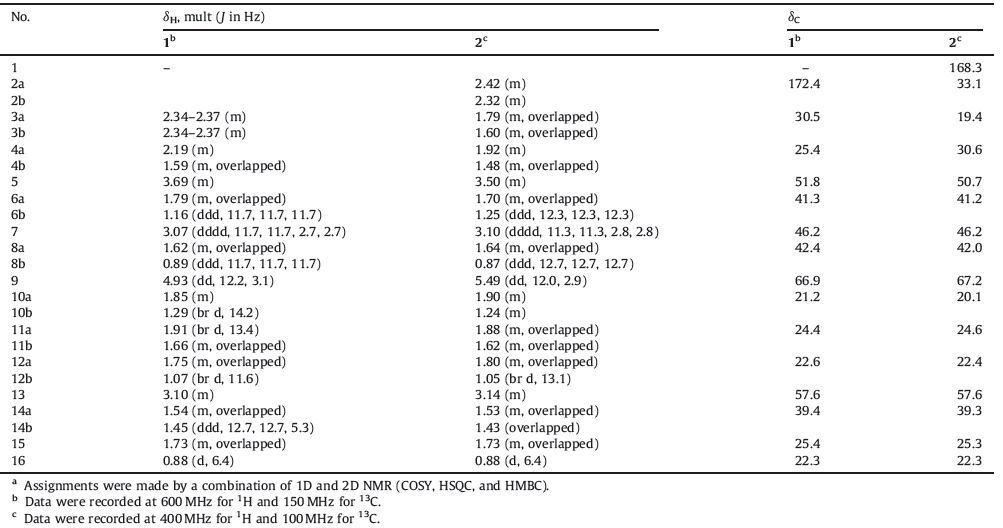
Palhinhaea (family Lycopodiaceae) is a small plant genus with only two species and two forma distributed in China [1]. Plants belonging to this genus have been well-documented to yield Lycopodium alkaloids [2-10]. P. cernua f. sikkimensis once could be widely found in southwestern China [1]; however,it has been harder and harder to collect due to the environmental change in the last decade. Previous phytochemical investigation on this plant led to the isolation of a number of serratane-type triterpenoids [11]. Nevertheless,alkaloidal constituents of this plant have never been studied yet. During our continuing research towards the discovery of novel bioactive alkaloids from club mosses [12, 13],the alkaloid-containing fraction of P. cernua f. sikkimensis was investigated. Reported herein are the isolation and structure elucidation of a novel [5/6/6/6]- (1) and five known [6/6/6/6]- cernuane-type alkaloids from the title plant (Fig. 1).

|
Download:
|
| Figure 1. The structures of compounds 1–6. | |
2. Experimental 2.1. General experimental procedures
Optical rotations were measured on an Autopol IV automatic polarimeter. UV and IR spectra were recorded on a Hitachi U- 2900E double-beam spectrophotometer and an Avatar 360 ESP FTIR spectrometer,respectively. ECD spectra were recorded on a JASCO-810 spectropolarimeter. NMR spectra were recorded on a Bruker Avance Ⅲ 400 MHz or 600 MHz spectrometer at 295 K. Chemical shifts are expressed inδ (ppm) and referenced to the residual solvent signals. ESIMSwere measured on an Agilent 1100 seriesmass spectrometers and HRESIMSweremeasured on an AB SCIEX Triple TOF 5600+ spectrometer. Semipreparative HPLC was performed on a Waters e2695 system with a Waters 2998 Photodiode Array Detector,a Waters 2424 ELSD,and a SunFire ODS column (250×10 mm2,5μm). Column chromatography (CC) was performed using silica gel (200-300mesh,Kang-Bi-Nuo Silysia Chemical Ltd.,Yantai,China),and Sephadex LH-20 (GE Healthcare Bio-Sciences AB,Uppsala,Sweden). Silica gel-precoated plates (GF254,0.25 mm,Kang-Bi-Nuo Silysia Chemical Ltd.,Yantai,China) were used for TLC detection. Spots were visualized using UV light (254 and/or 365 nm) and by spraying with Dragendorff reagent.
2.2. Plant materialThe whole plant of P. cernua f. sikkimensis (PCFS) was collected in September 2013 from Nanchuan,Chongqing of China. A voucher specimen (No. 20130907) was deposited at the Herbarium of the Department of Natural Products Chemistry,School of Pharmacy at Fudan University. The plant was identified by Mr. Qiu-Shi Tan (Chongqing Institute of Medicinal Plant Cultivation).
2.3. Extraction and isolationThe air-dried whole plant of PCFS (3.0 kg) was extracted with 90% MeOH (5×10 L) at room temperature,and the extract was partitioned between EtOAc and 3% tartaric acid. The water-soluble portion,adjusted to pH 9.0 with sat. Na2CO3,was partitioned with CHCl3. The CHCl3-soluble portion (4.5 g) was loaded on a silica gel column,eluted with a gradient CH2Cl2/MeOH (1:0-0:1,v/v) to afford fractions 1-6. Fraction 2 (570 mg) was subjected to silica gel CC (CH2Cl2/MeOH,30:1,v/v) and then purified by semi-preparative HPLC [MeOH-H2O (containing 0.05% Et2NH,v/v) 65:35,v/v; flow rate,3.0 mL/min] to furnish 2 (75.4 mg,tR = 21.4 min). Fraction 3 (120 mg) was separated by Sephadex LH-20 (MeOH) and further purified by semi-preparative HPLC [MeCN-H2O (containing 0.05% Et2NH,v/v) 40:60,v/v; flow rate,3.0 mL/min] to afford 1 (1.1 mg,tR = 14.1 min). Compound 3 (12.5 mg,tR = 9.6 min) was isolated from fraction 4 (80 mg) by semipreparative HPLC [MeOH-H2O (containing 0.05% Et2NH,v/v) 60:40,v/v; flow rate,3.0 mL/min]. Compound 4 (4.6 mg,tR = 11.3 min) was obtained from fraction 5 (70 mg) by using semi-preparative HPLC [MeCN-H2O (containing 0.05% Et2NH,v/v) 40:60,v/v; flow rate,3.0 mL/min]. Fraction 6 (300 mg) was separated by Sephadex LH-20 (CH2Cl2/MeOH,2:1,v/v) and further purified by semi-preparative HPLC [MeCN-H2O (containing 0.05% Et2NH,v/v) 20:80,v/v; flow rate,3.0 mL/min] to afford compounds 5 (2.7 mg,tR = 9.8 min) and 6 (3.4 mg,tR = 6.8 min).
Palcernuine (1): Colorless gum; [α]D 20-8 (c 0.1,MeOH); UV (MeOH)λmax(log ε) 201 (4.00) nm; ECD (c 1.21×10-3 mol/L,MeOH)λmax(Δε): 214 (-5.37) nm; IR (film) vmax (cm-1): 2922,2858,1673,1439,1364,1287,1053; 1H NMR and 13C NMR data,see Table 1; (+) ESIMS m/z 249 [M + H]+,519 [2M + Na] +; HRESIMS m/z 249.1963 [M + H]+ (calcd. for C15H25N2O,249.1961,Δ = +0.5 ppm).
|
|
Table 1 1H NMR and 13C NMR Data (δ ppm, in CDCl3) of 1a and 2. |
{kind=link}
Cernuine (2):Colorless gum; [α]D 20 -26 (c 0.1,MeOH) [lit. [α]D 20 -20.5 (c 0.1,MeOH)] [14]; UV (MeOH)λmax(log ε) 202 (3.89) nm; ECD (c 1.9×10-3 M,MeOH)λmax(Δε): 224 (-1.06) nm; IR (film) vmax (cm-1): 2922,2868,1641,1438,1346,1293,1220; 1H (CDCl3,400 MHz) and 13C (CDCl3 ,100 MHz) NMR data,see Table 1; 1H (CD3OD,400 MHz) and 13C (CD3OD,100 MHz) NMR data were identical to those in the literature [9]; (+) ESIMS m/z 263 [M+ H]+.
2α-Hydroxycernuine (4): Colorless gum; [α]D 20 -20 (c 0.1,MeOH) [lit. [α]D 20-22 (c 0.5,MeOH)] [5]; UV (MeOH)λmax(log ε) 203 (3.80) nm; ECD (c 1.8×10-3 mol/L,MeOH)λmax(Δε): 233 (-2.98) nm; 1H (CD3OD,400 MHz) NMR data were identical to those in the literature [5]; 1H NMR (C5D5N,600 MHz):δ 4.33 (dd,1H,J = 7.3,4.7 Hz,H-2β),2.00 (m,1H,H-3β),1.91 (dddd,1H,J = 12.3,9.4,7.3,4.5 Hz,H-3α),1.80 (m,2H,H-4),3.50 (m,1H,H-5α),1.32 (m,1H,H-6α),1.29 (ddd,1H,J = 12.6,12.6,12.6 Hz,H-6β),2.92 (m,1H,H-7α),1.41 (m,1H,H-8α),0.75 (ddd,1H,J = 12.0,12.0,12.0 Hz,H-8β),5.78 (dd,1H,J = 12.1,2.8 Hz,H-9β),1.96 (dddd,1H,J = 12.7,12.7,12.7,4.3 Hz,H-10α),1.19 (br d,1H,J = 12.7 Hz,H-10β),1.77 (m,1H,H-11α),1.50 (ddddd,1H,J = 13.1,13.1,13.1,3.9,3.9 Hz,H-11β),1.62 (dddd,1H,J = 12.5,12.5,12.5,3.9 Hz,H-12α),0.83 (br d,1H,J = 12.5 Hz,H-12β),2.97 (m,1H,H- 13b),1.37 (m,2H,H-14),1.54 (m,1H,H-15α),0.78 (d,3H,J = 6.4 Hz,Me-16); 1H NMR (CDCl3,400 MHz):δ 3.95 (dd,1H,J = 10.4,4.9 Hz,H-2β),2.04 (m,1H,H-3α),1.85 (m,1H,H-3β),2.00 (m,1H,H-4α),1.72 (m,1H,H-4β),3.70 (dddd,1H,J = 9.4,9.4,3.5,3.5 Hz,H-5),1.52 (m,1H,H-6α),1.46 (m,1H,H-6β),3.15 (m,1H,H- 7),1.59 (m,1H,H-8α),0.94 (ddd,1H,J = 12.1,12.1,12.1 Hz,H-8β),5.53 (dd,1H,J = 12.0,2.6 Hz,H-9),2.00 (m,1H,H-10α),1.21 (br d,1H,J = 12.8 Hz,H-10β),1.94 (m,1H,H-11α),1.68 (m,1H,H-11β),1.76 (m,1H,H-12α),1.08 (br d,1H,J = 12.7 Hz,H-12β),3.16 (m,1H,H-13),1.50 (m,2H,H-14),1.70 (m,1H,H-15),0.88 (d,3H,J = 6.4 Hz,Me-16); 13C NMR (CDCl3,100 MHz):δ 170.6 (C-1),68.2 (C-2),24.7 (C-3),25.3 (C-4),49.7 (C-5),40.0 (C-6),47.2 (C-7),41.5 (C-8),67.3 (C-9),20.7 (C-10),24.6 (C-11),22.3 (C-12),57.5 (C-13),38.8 (C-14),25.2 (C-15),22.2 (C-16); (+) ESIMS m/z 279 [M + H]+,579 [2M + Na] +. 1D-,2D-NMR,ESIMS,UV,ECD and/or HRESIMS spectra of compounds 1,2 and 4 are available in Supporting information.
2.4. General procedure for the ECD spectra calculationThe lowest energy conformation of 1 was optimized by using DFT method at B3LYP/6-31G(d,p) level with the PCM (MeOH) solvation model,and a set of thermodynamic data was generated. Each optimized structure was used for the TDDFT calculation at the same theoretic level and solvation model to generate a set of excitation energy and the corresponding rotational strength which were then plotted by using a Gaussian function to generate the calculated ECD spectra that was overlaid with experiment ECD spectra [15-17].
3. Results and discussionPalcernuine (1,1.1 mg) showed a pseudo-molecular ion peak at m/z 249.1963 in its HRESIMS,corresponding to the molecular formula C15H24N2O (calcd. for C15H25N2O,249.1961) with five degrees of unsaturation. In accordance with the molecular formula,fifteen well-resolved carbon signals were observed in the 13C NMR spectrum of 1 (Table 1). With the aid of DEPT and HSQC NMR experiments,they were classified as a methyl (δ 22.3),eight methylenes,five methines (four nitrogen-bearing atδ 46.2,51.8,57.6,and 66.9),and one carbonyl (δ 172.4) carbons. The 1H NMR spectrum of 1 displayed one secondary methyl group atδ 0.88 (δ ,3H,J = 6.4 Hz,Me-16) and four characteristic N-methine protons [9, 14, 18] atδ 3.69 (m,1H,H-5),3.07 (dddd,1H,J = 11.7,11.7,2.7,2.7 Hz,H-7),4.93 (dd,1H,J = 12.2,3.1 Hz,H-9),and 3.10 (m,1H,H- 13) (Table 1).
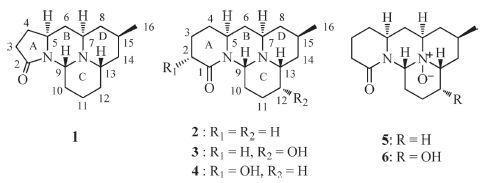
The above NMR data closely resembled those of cernuine (2) [9, 14, 18],a known cernuane-type alkaloid with a common [6/6/6/ 6]-tetracyclic ring system containing two nitrogen atoms previously isolated or detected from L. cernuum (=P. cernua) [7, 9, 14, 19, 20],and Lycopodium chinense [21]. Differing from 2,only fifteen carbons were found for alkaloid 1,with the loss of a CH2 group (Table 1),from both the 13CNMRand HRESIMS data. The COSY NMR spectrum of 1 revealed the presence of a long spin system of CH2-CH2-CH-CH2-CH-CH2-CH(CH3)-CH2-CH-CH2- CH2-CH2-CH,and the HMBC correlations of H2-3 (δ 2.34-2.37)/ C-2 (δ 172.4) and H-4α (δ 2.19)/C-2 confirmed that the carbonyl group was adjacent to C-3 (Fig. 2). Taken together,all these data suggested the presence of a five-membered γ-lactam ring in compound 1 instead of the d-lactam in 2. In good agreement with this assumption,the IR absorption band associated with C55O band stretching (1673 cm-1) in compound 1 was shifted to much higher frequency than that (1641 cm-1) of 2 due to the increased ring strain. Meanwhile,the chemical shifts of C-2 (corresponding to C-1 of 2),C-3,and C-4 in 1 substantially shifted from 2 to 11 ppm (Δδ1- 2 = 4.1,11.1 and-5.2 ppm,respectively) when compared with those of 2 (Table 1). Moreover,the chemical shift of the angular proton H-9 in 1 was significantly upfield shifted (Δδ =-0.56 ppm) when compared with 2 (Table 1). A reasonable explanation is that H-9 is located in the deshielding region of the carbonyl group at C-1 [14, 18, 19],and the distance between them is increased when ring A is a five-membered γ-lactam instead of the δ-lactam ring.

|
Download:
|
| Figure 2. COSY, key HMBC and NOE correlations of 1. | |
{kind=link}
The relative configuration of 1 was established by analysis of the coupling constants and the NOESY spectrum. From the large coupling constants (JH-5,H-6b = JH-6b,H-7 = JH-8b,H-15 = 11.7 Hz),H-5,H-7,and H-15 should all take axial positions. Furthermore,clear NOE correlations of H-5/H-7 and H-7/H-15 confirmed that these protons were a-oriented,whereas the NOE correlation between H- 9 and H-13,together with the lack of relations from H-9 and H-13 to H-5,H-7,and H-15,indicated that H-9 and H-13 were in the opposite orientations.
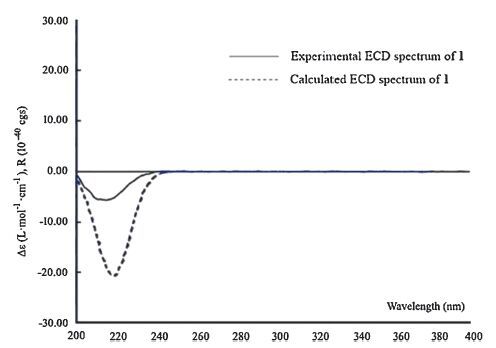
Finally,the ECD calculation [15-17] was employed to determine the absolute configuration of 1. The lowest energy conformation of compound1(Fig. 3)wasoptimizedbyusingDFTmethodatB3LYP/6- 31G(d,p) level. The optimized structures were then used for the time-dependent density functional theory (TDDFT) calculation. As a result,the calculated ECD spectrum of 1 was overlaid with the experimental ECD spectrum(Fig. 4),which unequivocally suggested an absolute configuration of 5S,7R,9S,13S,15S for compound 1. Indeed,both 1 and 2 were found to have the similar Cotton effect (negative) at ca. 220 nm (Supporting information),and the absolute configuration (5S,7R,9S,13S,15S) of the latter one has been undoubtedly determined by a total synthesis [22].

|
Download:
|
| Figure 3. The optimized structure of 1. | |
{kind=link}

|
Download:
|
| Figure 4. Calculated and experimental ECD spectra of 1 in MeOH. | |
{kind=link}
Comparing their spectroscopic data and physicochemical properties with those reported in the literature,the known ones were identified to be cernuine (2) [7, 9, 14, 18-21],lycocernuine (3) [9],2-hydroxycernuine (4) [5],cernuine N-oxide (5) [9],and lycocernuine N-oxide (6) [9],respectively. Among them,compound 4 was previously isolated from L. cernuum,but its relative configurations at C-2 and C-15 were ambiguous [5]. Based on a biogenetic consideration [10],the configuration of C-15 in 4 should be consistent with that of 1 and 2. Moreover,the proton coupling constants (JH-2,H-3 = 10.4,4.9 Hz,JH-8b,H-15 = 12.1 Hz,recorded in CDCl3) revealed that both H-2 and H-15 should be in axial positions (i.e.,b-orientation for H-2 and a-orientation for H-15). The NOESY spectrum measured in CDCl3 was obscure; however,a better set of NMR spectra (including 1H and NOESY) of 4 in C5D5N were then acquired. Clear NOE correlations of H-3α (δ 1.91)/H-5α (δ 3.50),H-5α/H-7α (d 2.92),and H-7α/H-15α (δ 1.54) as well as H-9β (δ 5.78)/H-13β (δ 2.97) were observed in the NOESY spectrum (measured in C5D5N,Supporting information) of 4. Thus,the structure of 2α-hydroxycernuine (4) was shown as depicted in Fig. 1.
To date,only ten naturally-occurring cernuane-type alkaloids have been reported,including cernuine (2) [7, 9, 14, 18-21],lycocernuine (3) [9],2α-hydroxycernuine (4) [5],cernuine N-oxide (5) [9],lycocernuine N-oxide (6) [9],palhinua A [2],anhydrolycocernuine [23],carolinianine [23],dihydrodeoxycernuine [23],and dihydrodeoxylycocernuine [23]. They all contain a common fused [6/6/6/6]-tetracyclic ring system. To the best of our knowledge,compound 1 is the first representative of cernuanetype alkaloids possessing an unprecedented [5/6/6/6]-tetracyclic ring system containing two nitrogen atoms.
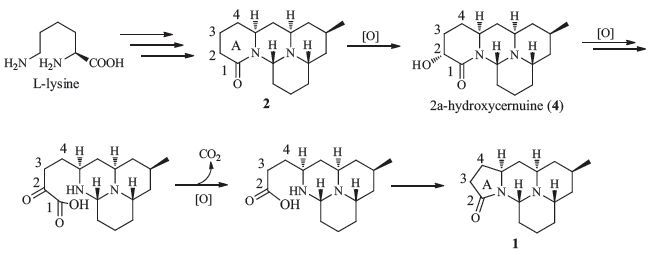
A biosynthetic pathway has been previously proposed for [6/6/ 6/6]-cernuane-type alkaloids from L-lysine (Scheme 1) [10]. Briefly,L-lysine could form cadaverine through decarboxylation. Cadaverine could be then transformed via 5-aminopentanal to afford D1-piperideine,which could further be coupled to acetonedicarboxylic acid (or its bisCoA ester) to yield piperidyl acetoacetate (4PAA) (or piperidyl acetoacetyl-CoA,4PAACoA). 4PAA/4PAACoA could be decarboxylated to form pelletierine. Accompanied by requisite decarboxylation,pelletierine and 4PAA/ 4PAACoA could then be coupled to form phlegmarine,the key intermediate toward cernuane-type alkaloids (e.g.,2). Obviously,this pathway does not directly predict the formation of the γ-lactam ring (ring A) in compound 1. Palcernuine (1) could be considered as a degradation product of 2 (Scheme 1). Cernuine (2) may be oxidized into 2-hydroxycernuine (4),which may undergo a series of oxidative reactions,including oxidative decarboxylation with concomitant ring cleavage,to afford 1 after lactam ring closure.

|
Download:
|
| Figure 1. Proposed biogenetic relationship between 1 and 2. | |
{kind=link}
The above isolates (except 1 due to its insufficient material) were evaluated for their cytotoxic effects against two human cancer cell lines (A-549 and NCI-H460) by MTT assay [24]. However,none of them showed significant cytotoxicity at a concentration of 50 mmol/L.
4. ConclusionDuring the first investigation on the alkaloidal constituents from the whole plant of P. cernua f. sikkimensis (PCFS),an unprecedented [5/6/6/6]-type (palcernuine,1) and five known [6/6/6/6]-type cernuane-type (2-6) alkaloids were obtained. As a small class of Lycopodium alkaloids,the cernuanes have only been reported from the following club mosses: P. cernua (=Lycopodium cernuum) [2, 5, 10],Lycopodium australianum [10],Lycopodium inundatum [10],Lycopodium carolinianum [10],Lycopodium carolinianum var. affine [10, 23],and Lycopodium chinense [10, 21]. Interestingly,all the known alkaloids (2-6) had previously been isolated from P. cernua. The results revealed a close chemotaxonomic relationship between PCFS and P. cernua. The isolated compounds (1-6) could stimulate future phytochemical/genomics studies of club mosses.
| [1] | Editor Committee for Flora of China of the Chinese Academy of Science. Flora of China,. (2004) pp69–73. |
| [2] | D.B. Zhang, J.J. Chen, L. Zhang, et al. , Bioactive alkaloids from Palhinhaea cernua. Phytochem. Lett. 10 (2014) 76–79. |
| [3] | L.B. Dong, Y.N. Wu, S.Z. Jiang, et al. , Isolation and complete structural assignment of Lycopodium alkaloid cernupalhine a: theoretical prediction and total synthesis validation. Org. Lett. 16 (2014) 2700–2703. |
| [4] | L.B. Dong, X. Gao, F. Liu, et al. , Isopalhinine A, a unique pentacyclic Lycopodium alkaloid from Palhinhaea cernua. Org. Lett. 15 (2013) 3570–3573. |
| [5] | S. Morel, I. Kerzaon, V. Roumy, et al. , A new cernuane-type alkaloid from Lycopodium cernuum. Biochem. Syst. Ecol. 45 (2012) 188–190. |
| [6] | L.B. Dong, J. Yang, J. He, et al. , Lycopalhine A, a novel sterically congested Lycopodium alkaloid with an unprecedented skeleton from Palhinhaea cernua. Chem. Commun. 48 (2012) 9038–9040. |
| [7] | F.W. Zhao, Q.Y. Sun, F.M. Yang, et al. , Lycopodium alkaloids from Palhinhaea cernua. J. Braz. Chem. Soc. 23 (2012) 349–354. |
| [8] | F.W. Zhao, Q.Y. Sun, F.M. Yang, et al. , Palhinine A, a novel alkaloid from Palhinhaea cernua. Org. Lett. 12 (2010) 3922–3925. |
| [9] | H. Morita, Y. Hirasawa, T. Shinzato, et al. , New phlegmarane-type, cernuane-type, and quinolizidine alkaloids from two species of Lycopodium. Tetrahedron 60 (2004) 7015–7023. |
| [10] | X.Q. Ma, D.R. Gang. The Lycopodium alkaloids. Nat. Prod. Rep 21 (2004) 752–772. |
| [11] | J. Yan, L. Sun, X. Zhang, et al. , Serratene triterpenoids from Palhinhaea cernua var. sikkimensis. Chem. Pharm. Bull. 57 (2009) 1381–1384. |
| [12] | Y. Tang, Y. Fu, J. Xiong, et al. , Casuarinines A-J, lycodine-type alkaloids from Lycopodiastrum casuarinoides. J. Nat. Prod. 76 (2013) 1475–1484. |
| [13] | Y. Tang, J. Xiong, J.F. Hu. Lycopodium alkaloids from Diphasiastrum complanatum. Nat. Prod. Commun. 10 (2015) 2091–2094. |
| [14] | W.A. Ayer, J.K. Jenkins, S. Valverde-Lopez, et al. , The alkaloids of Lycopodium cernuum L. I. the structures of cernuine and lycocernuine. Can. J. Chem 45 (1967) 433–443. |
| [15] | W.X. Wang, J.J. Zhu, Y.K. Zou, et al. , Trichotomone, a new cytotoxic dimeric abietane-derived diterpene from Clerodendrum trichotomum,. Tetrahedron Lett 54 (2013) 2548–2552. |
| [16] | J. Wang, W.Z. Zhai, Y.K. Zou, et al. , Eucalyptals D and E, new cytotoxic phloroglucinols from the fruits of Eucalyptus globulus and assignment of absolute configuration. Tetrahedron Lett. 53 (2012) 2654–2658. |
| [17] | M.J. Frisch, et al. , Gaussian, 09, Revision D. (2009) . |
| [18] | W.A. Ayer, J.K. Jenkins, K. Piers, et al. , The alkaloids of Lycopodium cernuum L. II. the stereochemistry of cernuine and lycocernuine. Can. J. Chem. 45 (1967) 445–450. |
| [19] | W.A. Ayer, J.K. Jenkins, S. Valverde-Lopez, et al. , The alkalcids of Lycopodium cernuum. Tetrahedron Lett. 5 (1964) 2201–2209. |
| [20] | L. Marion, R.H.F. Manske. The alkaloids of Lycopodium species. Can. J. Res 26B (1948) 1–2. |
| [21] | H. Morita, Y. Hirasawa, N. Yoshida, et al. , Senepodine A, a novel C22N2 alkaloid from Lycopodium chinense. Tetrahedron Lett. 42 (2001) 4199–4201. |
| [22] | Y. Nishikawa, M. Kitajima, H. Takayama. First asymmetric total syntheses of cernuane-type Lycopodium alkaloids, cernuine, and cermizine D. Org. Lett. 10 (2008) 1987–1990. |
| [23] | J.C. Braekman, L. Nyembo, P. Bourdoux, et al. , Alkaloid distribution in genus Lycopodium. Phytochemistry 13 (1974) 2519–2528. |
| [24] | S.B.Wu, Y.P. Ji, J.J. Zhu, et al. , Steroids fromthe leaves of Chinese Melia azedarach and their cytotoxic effects on human cancer cell lines. Steroids 74 (2009) 761–765. |