 2016, Vol. 27
2016, Vol. 27 



Foldamers are linear molecules that are driven by non-covalent forces to fold into discrete secondary structures [1, 2]. Switching between the folding and extending states of foldamers represents efficient control of the conformations of long organic molecules to produce artificial secondary structures, which may exhibit interesting materials or biological functions. Currently such switching has been realized by changing the solvent composition or temperature [1d] or outside stimuli, such as pH [3, 4], donor- acceptor interaction [5-7], guest complexation [8-12], metal coordination [13], light [14-19] and redox [20]. In 1964, Kosower and Cotter reported that bipyridinium (viologen) radical cations dimerized in water [21]. In the past decade, chemists have developed efficient methods for improving this inherently weak non-covalent interaction and thus utilizing it to construct advanced supramolecular architectures [22, 23]. Recently, our group reported that the folding-de-folding process of tetrathiafulvalene (TTF)-bipyridinium (BIPY2+)-alternating dynamic covalent polymers could be multiply tuned by conjugated radical cation dimerization and TTF-BIPY2+ donor-acceptor interaction [24, 25]. In this paper, we describe that two polymers, P1 and P2 (Fig. 1) , that consist of alternately incorporated naphthalene (NP) and BIPY2+ can form two pleated folding states which are driven by NP-BIPY2+ donor-acceptor interaction and BIPY-+ dimerization.

|
Download:
|
| Figure 1. The structures of polymers P1 and P2. | |
{kind=link}
2. Experimental
All solvents were dried before use according to standard procedures. All reagents were obtained from commercial suppliers and used without further purification. 1H NMR and 13C NMR spectra were recorded on a Bruker DPX 400 MHz spectrometer at 298 K, and the chemical shifts were referenced to the residual solvent peaks. Mass spectra (ESI and MALDI) were obtained on Shimadzu LCMS-2010EV or IonSpec 4.7 Tesla FTMS. Absorption spectra were measured on a Perkin-Elmer Lambda 750s UV-vis spectrometer. Luminescence spectra were measured on a Perkin- Elmer LS-= luminescence spectrometer. Compounds 2 [26], 5a and 5b [24], 6 [27] and 7.4PF6 [28] were prepared according to reported methods. The synthesis route of polymers P1 and P2 is showed in Scheme 1.

|
Download:
|
| Scheme. 1. Synthesis of polymers P1 and P2. | |
Compound 3: A solution of compounds 1 (0.19 g, 1.2 mmol) and 2 (1.5 g, 3.6 mmol) in toluene (10 mL) and Et3N (2 mL) was stirred and refluxed under argon for 12 h, and then the mixture was concentrated under reduced pressure. After workup, the resulting slurry was subjected to column chromatography (MeOH/CH2Cl2 1:20) to give compound 3 as a colorless oil (0.35 g, 45%). 1H NMR (400 MHz, CDCl3): 7.61 (d, 2H, J = 8.8 Hz), 7.17-7.13 (m, 2H), 7.09 (s, 2H), 4.25-4.20 (m, 4H), 4.15 (s, 4H), 3.94-3.88 (m, 4H), 3.79- 3.61 (m, 30H). 13C NMR (100 MHz, CDCl3): 170.9, 154.4, 126.8, 125.1, 114.6, 105.7, 71.0, 70.9, 70.8, 70.7, 69.8, 68.6, 68.0, 51.8. MS (ESI): m/z 657.3 [M + H]+. HRMS (ESI): Calcd. for C32H49O14 [M + H]+: 657.3122. Found: 657.3118.
Compound 4: A solution of compound 3 (0.15 g, 0.23 mmol) and hydrazine monohydrate (1.00 mL, 98%) in methanol (10 mL) was stirred at room temperature for 2 h and then evaporated with a rotavapor. The resulting residue was triturated with CH2Cl2 (50 mL) and the solution washed with water (50 mL ± 2) and brine (50 mL), and then dried over anhydrous MgSO4. Upon removal of the solvent under reduced pressure, compound 4 was obtained as a white solid (0.15 g, 98%). 1H NMR (400 MHz, CD3CN): d 8.17 (br, 2H), 7.68 (d, 2H, J = 8.9 Hz), 7.22 (s, 2H), 7.14-7.08 (m, 2H), 4.26-4.11 (m, 4H), 3.93 (s, 4H), 3.88-3.78 (m, 4H), 3.70-3.50 (m, 24H), 2.17 (br, 4H). 13C NMR (100 MHz, CD3CN): 170.0, 156.2, 130.7, 129.1, 119.9, 108.0, 71.8, 71.3, 71.2, 71.1, 70.7, 70.6, 70.2, 68.4. MS (ESI): m/z 657.3 [M + H]+. HRMS (ESI): Calcd. for C30H49N4O12 [M + H]+: 657.3347. Found: 657.3331.
Polymer P1: Compound 5a (30 mg, 0.046 mmol) was dissolved in acetonitrile (5 mL). To the solution was added dropwise a solution of compound 4 (30 mg, 0.046 mmol) in acetonitrile (5 mL). The mixture was stirred at room temperature for 18 h and then the solvent was removed with a rotavapor. The resulting residue was suspended in dichloromethane and ether (1:1, 0.5 mL). The solid formed was filtrated off and washed thoroughly with ether, and dried in vacuo to give polymer P1 as a dark brown solid (54 mg, 92%).
Polymer P2: Polymers P2 were prepared as a dark brown solid from the reaction of 5b and 4 in 91% yield according to the procedure described for polymer P1.
3. Results and discussionPolymers P1 and P2 were prepared from the condensation reactions of 4 with 5a or 5b through the formation of hydrazone bonds in acetonitrile (Scheme 1) [24, 25]. After the reactions reached equilibrium, the signal of the hydrogen atom of the aldehyde groups disappeared completely in the 1H NMR spectra in acetonitrile-d3. Considering the sensitivity of the 1H NMR spectroscopy, we assumed that this observation reflected that at least 95% of the dialdehyde and the dihydrazine precursors were consumed to produce the dynamic covalent polymers and thus estimated the degree of polymerization of the two polymers to be ≥10. Gel permeation chromatography (GPC) experiments in acetonitrile and N, N-dimethylformamide were also performed for the two polymers, which did not afford useful results for the determination of the degree of polymerization. Both polymers were soluble in acetonitrile and thus their conformations were investigated in this solvent.
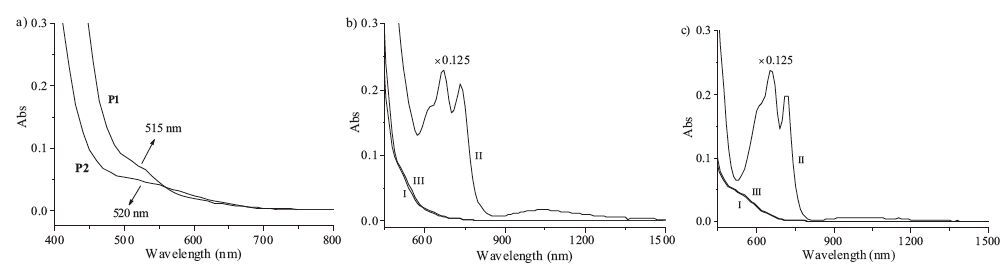
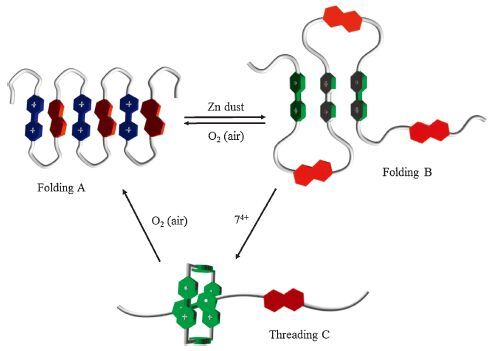
UV-vis spectra were first recorded for P1 and P2 in acetonitrile. Both polymers exhibited a broad absorption band centered around 515 and 520 nm, respectively (Fig. 2a), which supported the formation of the charge-transfer (CT) complexation between the electron-donating NP units and the electron-deficient BIPY2+ units. The molar absorption coefficient (ε) of P1 and P2 were calculated to be ca. 335 and 240 L/(mol cm), respectively. The absorption was linearly correlated to the concentration within the range of 0.11- 1.5 mmol/L, which indicated that the absorption band was caused by intramolecular donor-acceptor interaction. Thus, the backbone of both polymers should produce a pleated folded conformation (Folding A), as shown in Fig. 3.

|
Download:
|
| Figure 2. The UV–vis absorption spectra in acetonitrile at 25 8C of (a) polymers P1 and P2 ([BIPY2+] = 0.2 mmol/L), (b) polymer P1 ([BIPY2+] = [NP] = 0.2 mmol/L), and (c) polymer P2 ([BIPY2+] = [NP] = 0.2 mmol/L). I: Recorded for the original solution; Ⅱ: Recorded after excess of zinc dust was added; Ⅲ: Recorded after the solutions were exposed to air for 2 h. | |
{kind=link}

|
Download:
|
| Figure 3. Schematic diagram of the proposed pleated foldamers and threading complexes of polymers P1 and P2 driven by the donor-acceptor interaction and the BIPY·+ radical cation dimerization. | |
{kind=link}
Adding 5.0 equiv. of electron-rich macrocyclic polyether bis- 1, 5-dinaphtho [38] crown-10 (6) (Fig. 4) , which was relative to the concentration of the BIPY2+ units of the polymers, to the solution of P1 or P2 in acetonitrile did not cause observable change in the CT absorption band of both polymers. This result indicated that the crown ether could not break the intramolecular donor-acceptor interaction of the polymers by forming threading complexes with the BIPY2+ units of the polymers, supporting the high stability of this pleated folded conformation. Adding 2.0 equiv. of electrondeficient cyclobis(paraquat--henylene) cyclophane (7.4PF6) (Fig. 4) , which was relative to the concentration of the NP units of the polymers, to the solution of P1 or P2 in acetonitrile also caused no discernible increase of the CT absorption band even after keeping at room temperature for 15 days. It was established that the tetracationic ring of 7.4PF6 is not able to move over the BIPY2+ units incorporated into linear molecules owing to electrostatic repulsion [29]. This observation supported that this tetracationic cyclophane did not either form encapsulation complexation with the NP units of the polymers at their ends. This encapsulation, if occurred [24, 25], was expected to cause an enhancement of the CT absorption and de-folding of these end-appended NP units (Folding A).

|
Download:
|
| Figure 4. The structure of compounds 6 and 7.4PF6. | |
{kind=link}
Then we used the UV-vis absorption spectroscopy to investigate the conformations of the two polymers in acetonitrile after the BIPY2+ units were reduced to radical cations with excess of zinc dust (Fig. 2b and c). For both polymers, the spectra exhibited strong absorption bands centered around 670 and 650 nm, respectively, which corresponded to the monomeric radical cation BIPY·+ [22-25]. The spectra also displayed a broad absorption band around 1040 and 980 nm, respectively, which was typical of the radical cation dimer (BIPY·+)2 [22-25]. This absorption band was not observed for both 5a and 5b of the identical concentration after they were reduced to radical cation. Moreover, the intensity of these two absorption bands was also linearly correlated with the concentration within the investigated concentration range (0.10- 1.5 mmol/L) and thus could be attributed to the intramolecular stacking of the (BIPY·+)2 units and supported the formation of another pleated folded state [24] (Folding B, Fig. 3) . In both folding states, the driving forces came from dynamic non-covalent interactions. Thus, the flexible de-folding conformation could not be excluded. Nevertheless, the two folding states should be energetically favored. The folding also reduced the flexibility of the backbone. Thus, the stacking between the neighboring aromatic units might be promoted to each other. However, multivalence, if any, might be quite weak.
Electron paramagnetic resonance (EPR) experiments were further carried out for polymers P1 and P2 and control compounds 5a/5b in acetonitrile after their BIPY2+ units were reduced to (BIPY·+) Compared with that of 5a and 5b, whose BIPY-+ units did not dimerize, the EPR signal of the two polymers, with the identical BIPY concentration, were substantially weaker (Fig. 5) . The observation should be ascribed to the dimerization of most part of BIPY-+ units of the polymers. These results could be regarded as another evidence supporting the construction of Folding B, because the (BIPY·+)2 units were diamagnetic and thus EPR-silent. Upon exposing to air for 2 h, the BIPY-+ radical cations of the two polymers were oxidized back to the dicationic state. The pleated Folding A was recovered, as evidenced by the regeneration of the CT band (Fig. 2b and c).

|
Download:
|
| Figure 5. EPR spectra of the solution of (a) 5a and P1, (b) 5b and P2 in acetonitrile containing excess of zinc dust at 25 8C ([BIPY] = 0.2 mmol/L). | |
{kind=link}
It has been reported that the 72(·+) could form a stable inclusion complex with the BIPY·+ unit of linear molecules [30]. We thus conjectured that 72(·+) might also encapsulate the BIPY·+ units of polymers P1 and P2 to cause the de-folding of their pleated conformation (Folding B). The absorption spectra were then recorded for the solutions of the two polymers in acetonitrile ([BIPY] = 0.2 mmol/L). At the reduction state of both polymers, adding 1.0 equiv. of 72(·+), relative to [BIPY] of the polymers, did cause substantial enhancement of the absorption of the (BIPY·+)2 dimers of the polymers. At the concentration of 1.0 mmol/L, the absorption spectrum of 72(·+) did not give rise to any absorption band of the (BIPY·+)2 unit. Thus, the above absorption enhancement should support the formation of more stable threading complexes (Threading C, Fig. 3) as a result of the encapsulation of the BIPY·+ units of the polymers by the 72(·+) ring, which led to the formation of the de-folding conformation. When the solutions were exposed to air, the radical cations could be oxidized to produce BIPY2+, leading to the recovery of Folding A. Cyclic voltammograms were also recorded for both polymers in acetonitrile, which displayed only very broad, unassignable peaks probably due to the strong stacking of the radical cation units.
4. ConclusionIn conclusion, we have demonstrated that naphthalene- and bipyridinium-alternately incorporated polymers P1 and P2 can form two different pleated foldamers, which are stabilized by intramolecular donor-acceptor interaction and conjugated radical cation dimerization. The two folded conformations can switch into each other by reversible redox of the electron-deficient bipyridinium units. The results showed that intermolecular noncovalent interactions function overwhelmingly at relatively low concentrations to enable the control of a regular conformation for linear polymers. In the future, we will design linear polymers by changing the ratio of the donor and acceptor units. In this way, more advanced pleated secondary structures may be created in the three-dimensional space and intermolecular interactions may be imposed to lead to new methodologies for conformational regulation.
AcknowledgmentThis work was financially supported by The Ministry of Science and Technology of China (No. 2013CB834501) , The Science and Technology Commission of Shanghai Municipality (No. 13M1400200) , The Ministry of Education of China research fund for the doctoral program, and the National Natural Science Foundation of China (Nos. 21432004 and 21472023) .
| [1] |
(a) S. Hecht, I. Huc (Eds.), Foldamers: Structure, Properties and Applications, Wiley-VCH, Weinheim, Germany, 2007, p. 456; (b) S.H. Gellman, Foldamers: a manifesto, Acc. Chem. Res. 31 (1998) 173-180;v (c) D.J. Hill, M.J. Mio, R.B. Prince, T.S. Hughes, J.S. Moore, A field guide to foldamers, Chem. Rev. 101 (2001) 3893-4012; (d) B. Gong, Crescent oligoamides: from acyclic "Macrocycles" to folding nanotubes, Chem. Eur. J. 7 (2001) 4336-4342; (e) Z.T. Li, Supramolecular chemistry: from aromatic foldamers to solution-phase supramolecular organic frameworks, Beilstein J. Org. Chem. 11 (2015) 2057-2071. |
| [2] |
(a) Z.T. Li, J.L. Hou, C. Li, Peptide mimics by linear arylamides: a structural and functional diversity test, Acc. Chem. Res. 41 (2008) 1343-1353; (b) B. Gong, Hollow crescents, helices, and macrocycles from enforced folding and folding-assisted macrocyclization, Acc. Chem. Res. 41 (2008) 1376-1386; (c) X. Li, Y.D. Wu, D. Yang, a-Aminoxy acids: new possibilities from foldamers to anion receptors and channels, Acc. Chem. Res. 41 (2008) 1428-1438; (d) G.N. Tew, R.W. Scott, M.L. Klein, W.F. De Grado, De Novo design of antimicrobial polymers, foldamers, and small molecules: from discovery to practical applications, Acc. Chem. Res. 43 (2010) 30-39; (e) D.W. Zhang, X. Zhao, J.L. Hou, Z.T. Li, Aromatic amide foldamers: structures, properties, and functions, Chem. Rev. 112 (2012) 5271-5376; (f) H. Fu, Y. Liu, H. Zeng, Shape-persistent H-bonded macrocyclic aromatic pentamers, Chem. Commun. 49 (2013) 4127-4144; (g) Y. Zhao, K. Cho, L. Widanapathirana, S. Zhang, Conformationally controlled oligocholate membrane transporters: learning through water play, Acc. Chem. Res. 46 (2013) 2763-2772; (h) D.W. Zhang, W.K. Wang, Z.T. Li, Hydrogen-bonding-driven aromatic foldamers: their structural and functional evolution, Chem. Rec. 15 (2015) 233-251. |
| [3] | E. Kolomiets, V. Berl, J.M. Lehn. Chirality induction and protonation-induced molecular motions in helical molecular strands. Chem. Eur. J. 13 (2007) 5466–5479. |
| [4] | H.Y. Hu, J.F. Xiang, Y. Yang, C.F. Chen. Chiral induction in phenanthroline-derived oligoamide foldamers: an acid-and base-controllable switch in helical molecular strands. Org. Lett. 10 (2008) 1275–1278. |
| [5] | R.S. Lokey, B.L. Iverson. Synthetic molecules that fold into a pleated secondary structure in solution. Nature 375 (1995) 303–305. |
| [6] |
(a) S. Ghosh, S. Ramakrishnan, Aromatic donor-acceptor charge-transfer and metal-ion-complexation-assisted folding of a synthetic polymer, Angew. Chem. Int. Ed. 43 (2004) 3264-3268; (b) S. Ghosh, S. Ramakrishnan, Small-molecule-induced folding of a synthetic polymer, Angew. Chem. Int. Ed. 44 (2005) 5441-5447; (c) K. Liu, X. Zheng, A.Z. Samuel, et al., Stretching single polymer chains of donor-acceptor foldamers: toward the quantitative study on the extent of folding, Langmuir 29 (2013) 14438-14443. |
| [7] | X. Zhao, M.X. Jia, X.K. Jiang, et al. Zipper-featured d-peptide foldamers driven by donor-acceptor interaction. Design, synthesis, and characterization. J. Org. Chem. 69 (2004) 270–279. |
| [8] | Y.X. Xu, X. Zhao, X.K. Jiang, Z.T. Li. Folding of aromatic amide-based oligomers induced by benzene-1, 3, 5-tricarboxylate anion in DMSO. J. Org. Chem. 74 (2009) 7267–7273. |
| [9] | Y. Wang, J. Xiang, H. Jiang. Halide-guided oligo(aryl-triazole-amide)s foldamers: receptors for multiple halide ions. Chem. Eur. J. 17 (2011) 613–619. |
| [10] | K.P. McDonald, Y. Hua, S. Lee, A.H. Flood. Shape persistence delivers lock-and-key chloride binding in triazolophanes. Chem. Commun. 48 (2012) 5065–5075. |
| [11] | C. Sun, C. Ren, Y. Wei, B. Qin, H. Zeng. Patterned recognition of amines and ammonium ions by a stimuli-responsive foldamer-based hexameric oligophenol host. Chem. Commun. 49 (2013) 5307–5309. |
| [12] | L. Cera, C.A. Schalley. Stimuli-induced folding cascade of a linear oligomeric guest chain programmed through cucurbit[n]uril self-sorting (n =, 6, 7, 8). Chem. Sci. 5 (2014) 2560–2567. |
| [13] | R.M. Meudtner, M. Ostermeier, R. Goddard, C. Limberg, S. Hecht, Multifunctional "Clickates" as versatile extended heteroaromatic building blocks: efficient synthesis via click chemistry, conformational preferences, and metal coordination. Chem. Eur. J. 13 (2007) 9834–9840. |
| [14] | Z. Yu, S. Hecht. Reversible and quantitative denaturation of amphiphilic oligo(azobenzene) foldamers. Angew. Chem. Int. Ed. 50 (2011) 1640–1643. |
| [15] | A. Khan, C. Kaiser, S. Hecht. Prototype of a photoswitchable foldamer. Angew. Chem. Int. Ed. 45 (2006) 1878–1881. |
| [16] | Y. Wang, F. Bie, H. Jiang. Controlling binding affinities for anions by a photoswitchable foldamer. Org. Lett. 12 (2010) 3630–3633. |
| [17] | S. Lee, Y. Hua, A .H. Flood, b-sheet-like hydrogen bonds interlock the helical turns of a photoswitchable foldamer to enhance the binding and release of chloride. J. Org. Chem. 79 (2014) 8383–8396. |
| [18] | Y. Hua, A.H. Flood. Flipping the switch on chloride concentrations with a lightactive foldamer. J. Am. Chem. Soc. 132 (2010) 12838–12840. |
| [19] |
(a) Z. Yu, S. Hecht, Cooperative switching events in azobenzene foldamer denaturation, Chem. Eur. J. 18 (2012) 10519-10524; (b) Z. Yu, S. Hecht, Control over unfolding pathways by localizing photoisomerization events within heterosequence oligoazobenzene foldamers, Angew. Chem. Int. Ed. 52 (2013) 13740-13744; (c) Z. Yu, S. Weidner, T. Risse, S. Hecht, The role of statistics and microenvironment for the photoresponse in multi-switch architectures: the case of photoswitchable oligoazobenzene foldamers, Chem. Sci. 4 (2013) 4156-4167. |
| [20] | D. Siebler, M. Linseis, T. Gasi, et al. , Oligonuclear ferrocene amides: mixed-valent peptides and potential redox-switchable foldamers. Chem. Eur. J. 17 (2011) 4540–4551. |
| [21] | E. Kosower, J. Cotter, Stable free radicals. Ⅱ. The reduction of 1-methyl-4-cyanopyridinium ion to methylviologen cation radical, J. Am. Chem. Soc. 86 (1964) 5524-5527. |
| [22] |
(a) D.W. Zhang, J. Tian, L. Chen, L. Zhang, Z.T. Li, Dimerization of conjugated radical cations: an emerging non-covalent interaction for self-assembly, Chem. Asian J. 10 (2015) 56-68; (b) L. Chen, Y.C. Zhang, W.K. Wang, et al., Conjugated radical cation dimerizationdriven generation of supramolecular architectures, Chin. Chem. Lett. 26 (2015) 811-816; (c) H. Wang, D.W. Zhang, X. Zhao, Z.T. Li, Supramolecular organic frameworks (SOFs): water-phase periodic porous self-assembled architectures, Acta Chim. Sin. 73 (2015) 471-479; (d) Z.T. Li, Supramolecular chemistry: from aromatic foldamers to solution-phase supramolecular organic frameworks, Beilstein J. Org. Chem. 11 (2015) 2057-2071; (e) T.Q. Wan, Z.T. Li, From supramolecular polymers to supramolecular organic frameworks: engineering the periodicity of solution-phase self-assembled architectures, Imag. Sci. Photochem. 33 (2015) 3-14; (f) L. Zhang, T.Y. Zhou, J. Tian, et al., A two-dimensional single-layer supramolecular organic framework that is driven by viologen radical cation dimerization and further promoted by cucurbit[8]uril, Polym. Chem. 5 (2014) 4715-4721. |
| [23] | Y. Wang, M. Frasconi, W.G. Liu, et al. , Folding of oligoviologens induced by radical-radical interactions. J. Am. Chem. Soc. 137 (2015) 876–885. |
| [24] | L. Chen, H. Wang, D.W. Zhang, Y. Zhou, Z.T. Li. Quadruple switching on pleated foldamers of tetrathiafulvalene-bipyridinium-alternating dynamic covalent polymers. Angew. Chem. Int. Ed. 54 (2015) 4028–4031. |
| [25] | Y.C. Zhang, D.W. Zhang, H. Wang, Y. Zhou, Z.T. Li. Bipyridinium radical cation dimerization-driven polymeric pleated foldamers and a homoduplex that undergo ion-tuned interconversion. Polym. Chem. 6 (2015) 4404–4408. |
| [26] | B. Chen, U. Baumeister, G. Pelzl, et al. , Carbohydrate rod conjugates: ternary rod coil molecules forming complex liquid crystal structures. J. Am. Chem. Soc. 127 (2005) 16578–16591. |
| [27] | P.R. Ashton, E.J.T. Chrystal, J.P. Mathias, et al. , Complexation of diquat and paraquat by macrocyclic polyethers incorporating two dibydroxynaphthalene residues. Tetrahedron Lett. 28 (1987) 6367–6370. |
| [28] | P.L. Anelli, P.R. Ashton, R. Ballardini, et al., Molecular meccano. 1. [2]rotaxanes and a [2]catenane made to order, J. Am. Chem. Soc. 114 (1992) 193-218. |
| [29] |
(a) M. Hadeh, A.C. Fahrenbach, S. Basu, et al., Electrostatic barriers in rotaxanes and pseudorotaxanes, Chem. Eur. J. 17 (2011) 6076-6087; (b) H. Li, Y.L. Zhao, A.C. Fahrenbach, et al., Degenerate [2]rotaxanes with electrostatic barriers, Org. Biomol. Chem. 9 (2011) 2240-2250. |
| [30] | H. Li, Z. Zhu, A.C. Fahrenbach, et al. , Mechanical bond-induced radical stabilization. J. Am. Chem. Soc. 135 (2013) 456–467. |