 2016, Vol. 27
2016, Vol. 27 



b School of Pharmacy, Second Military Medical University, Shanghai 200433, China ;
c Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
In recent years,the population of immunocompromised individuals,such as AIDS and organ transplants recipient,is increasing. This fact makes the invasive fungal infection become a global threat to human health [1, 2]. Among the drugs to treat fungal infection,triazole derivatives as potent and safe antifungal agents have attracted attention for a long time [3, 4]. Fluconazole, voriconazole,and itraconazole (Fig. 1) have been widely used in clinical therapy. But some impediments of these drugs still remain to be resolved. Firstly,with the extensive use of triazole antifungal drugs,the rate of drug resistance mutation is also increasing gradually [5, 6]. Secondly,the poor water-solubility and drug-drug interactions are a common problem of triazole antifungal drugs [7]. Therefore,the discovery and development of novel antifungal agents for clinical treatment has important realistic meanings.

|
Download:
|
| Fig. 1. Chemical structure of fluconazole, voriconazole, albaconazole, and itraconazole. | |
Albaconazole (Fig. 1) is one of the most potent antifungal agents reported,which has potent activity against many fungi such as Candida spp and Aspergillus spp [8]. A quinazolinone unit rather than a pyrimidine unit of voriconazole is the structure characteristic of albaconazole. Literatures have disclosed the important interaction between the carbonyl of quinazolinone unit with the His310 of CYP51,the target enzyme of triazole antifungal agents [9]. Our lab also has done some work in this area [10, 11]. For example,the pyridine-substituted analogues of itraconazole,some of which showed good activities against pathogenic fungi. Unfortunately,all of those derivatives have poor water-solubility. Our previous studies found that the triazole derivatives containing 4,5,6,7-tetrahydrothiazolo[5,4-c]pyridine motif have good activities against Candida spp in vitro [12]. Compound 1 (Fig. 2) has the most potent activity among those derivatives. What is important is that the disulfate salt of compound 1 has good water-solubility,which can be given through intravenous injection.

|
Download:
|
| Fig. 2. The design of title compounds. | |
Herein,we designed and synthesized two series of triazole derivatives featuring 5,6-dihydro-4H-pyrrolo[3,4-d]thiazol-4-one moiety 2 and 4,5-dihydro-6H-pyrrolo[3,4-d]thiazol-6-one moiety 3 (Fig. 2) on the basis of the structure characteristics of 1 and albaconazole. We hypothesized that the carbonyl of γ-lactam could interact with the His310 of CYP51,which can enhance the antifungal activity.
2. ExperimentalThe synthetic route of title compounds 2A-D is outlined in Scheme 1. The reaction of compound 4 with ethylmagnesium bromide yielded 5. Then compound 5 reacted with CuBr2 to give 6, which underwent annelation to obtain 7. Compound 7 underwent a Sandemyer reaction and bromination by NBS sequentially to afford 9. The key intermediate 9 reacted with compound 10 [13] to give 11, which underwent a hydrolysis and condensation to obtain compound 2A. Compounds 2B-D were obtained by treating 2A with different boronic acid pinacol ester 12B-D in 1,4-dioxane and water.

|
Download:
|
| Scheme. 1. Synthetic route for title compounds 2A–D. Reagents and conditions: (a) ethylmagnesium bromide, THF, -78 ℃ then -10 ℃, 2 h; (b) CuBr2, EtOAc/CHCl3, reflux,12 h; (c) thiourea, EtOH, reflux 2 h then r.t., 12 h; (d) tBuONO, CuBr2, CH3CN, r.t. then 60 ℃, 6 h; (e) NBS, BPO, CCl4, 60 ℃, 12 h; (f) 9, K2CO3, CH3CN, 0 ℃ then r.t. overnight; (g) NaOH, THF, MeOH, 0 ℃ then r.t. overnight; (h) EDCI, HOBt, TEA, CH2Cl2, 0 ℃ then r.t. 2 h; (i) 12B–D, Pd[P(Ph3)4], Cs2CO3, 1, 4-dioxane/H2O,70 ℃, 12 h. | |
The synthetic route of compounds 3A-H is showed in Scheme 2. Commercially available compound ethyl 2-amino-4- methylthiazole-5-carboxylate(13) underwenta Sandemyer reaction to give compound 14,which was treated with different boronic acid pinacol ester 15B-H in 1,4-dioxane and water,giving compounds 16B-H. Compounds 16B-H and 14 reacted with NBS in the solution of CCl4 to yield compounds 17A-H. With the important intermediates 17A-H in hand,the title compounds 3A-H can be formed easily through the similar procedures outlined in Scheme 1.

|
Download:
|
| Scheme. 2. Synthetic route for title compounds 3A–H. Reagents and conditions: (a) tBuONO, CuBr2, DMF, CH3CN, 0 ℃ then 60 ℃, 1 h; (b) 15B–H, Cs2CO3, Pd(PPh3)4, 1, 4-dioxane/H2O, 80 ℃, 12 h; (c) NBS, AIBN, CCl4, r.t., overnight; (d) K2CO3, CH3CN, 0 ℃ then r.t. overnight; (e) NaOH, MeOH, THF, 0 ℃ then r.t. 8 h; (f) EDCI, HOBt, TEA, 0 ℃ then r.t. 2 h. | |
In vitro minimal inhibitory concentrations (MIC) of the title compounds were determined using the serial dilution method in 96-well microtest plates. The six pathogenic fungi were obtained from ATCC or clinic isolates. The MIC determination was performed according to Clinical and Laboratory Standards Institute (CLSI) guidelines [14, 15]. All of test compounds were dissolved in DMSO serially diluted in growth medium.
3. Results and discussionAs outlined in the Schemes 1 and 2,we can obtain the title compounds 2A-D and 3A-H smoothly and efficiently. Their structures were confirmed by 1H NMR,13C NMR and HRMS (Supplementary data). The MIC80 values of the title compounds are showed in Table 1. Our previous experiments showed that DMSO has no influence on the growth of the fungi tested under the test conditions. So,the results of blank control experiments (DMSO only) are not showed in this table.
|
|
Table 1 In vitro antifungal activity of the title compounds (MIC80, mg/mL). |
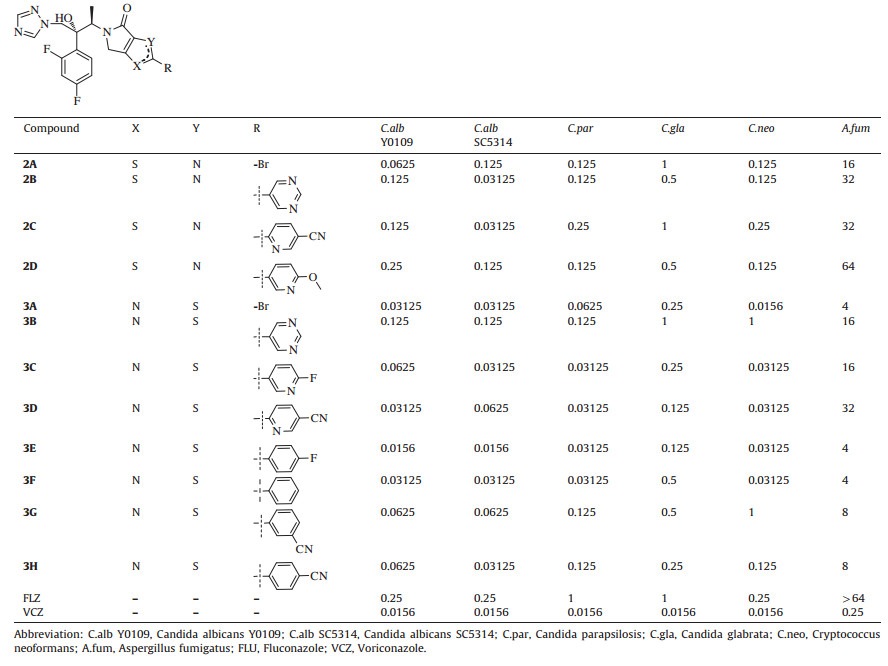
All the target compounds showed stronger activity against Candida spp than fluconazole. And especially some compounds (3D,3E,and 3F) have comparative MIC80 values against all the tested fungi as voriconazole except for Candida glabrata and Aspergillus fumigatus. What is important is that some compounds of series 3 (3A,3C,3D,3E,3F) showed good activities against Cryptococcus neoformans which has been increasing prevalence during the last few decades [16]. In the overall view,the compounds of series 3 showed more potent activity than compounds of series 2. Furthermore,the simple bromo-substituted compounds (2A and 3A) demonstrated stronger activity against all the six fungi than other aromatic substituted compounds (2B-D and 3B-H) except for 3E. In addition,the different aromatic rings (2B and 2C,3D and 3F) have little influence on the activity,which can be explained by the fact that the conformation of the ligand tunnels is compatible with different ligands [17].
The docking model of representative compound 2A and fungal CYP51 [18] was investigated using the GOLD Suite v5.0 software package (Fig. 3). Like the other triazole compounds,the 4-N of the triazole unit has a van Der Waals interaction with the heme iron. In addition,we can see that the 5,6-dihydro-4H-pyrrolo[3,4- d]thiazol-4-one unit was located in the hydrophobic pocket composed of Phe-241,Phe-236,Ile-139 and Try-140. Furthermore, the bromine atom of 2A have a halogen bond with Ser-382 (not showed),which can explain the potent activities of 2A and 3A.

|
Download:
|
| Fig. 3. The docking model for compound 2A and CYP51. | |
4. Conclusion
In summary,a series of novel triazole derivatives containing γ-lactam were designed and synthesized. Their in vitro antifungal activities against six pathogenic fungi were evaluated. According to the results,the R group of the thiazole ring was well tolerated in vitro. For example,both the pyridyl-substituted compound 3D and phenyl-substituted compound 3E exhibited good activity against the Candida spp and Cryptococcus neoformans tested. However,the activities of these novel triazole derivatives against Aspergillus fumigatus are moderate and the further studies to improve activity against Aspergillus fumigatus are ongoing in our lab. In addition,the cytotoxicities and metabolic properties of these novel triazole derivatives will be determined in due time.
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2016.01.040
| [1] | D. Armstrong-James, G. Meintjes, G.D. Brown. A neglected epidemic:fungal infections in HIV/AIDS. Trends Microbiol 22 (2014) 120–127 |
| [2] | M.C. Fisher, D.A. Heck, C.J. Briggs, et al. Emerging fungal threats to animal, plant and ecosystem health. Nature 484 (2012) 186–194 |
| [3] | S.S. Sandhu, H. Shukla, R.P. Aharwal, S. Kumar, S. Shukla. Antifungal azole derivatives and their pharmacological potential:prospects & retrospects. Nat. Prod. J. 4 (2014) 140–152 |
| [4] | V.K. Gupta, A.K. Sharma, R. Sharma, S. Diwan, S. Saini. Azoles as effective antifungal agents:trends, scope and relevance. Nat. Prod. J. 4 (2014) 82–92 |
| [5] | M.A. Pfaller. Antifungal drug resistance:mechanisms, epidemiology, and consequences for treatment. Am. J. Med 125 (2012) S3–S13 |
| [6] | J.B. Anderson. Evolution of antifungal-drug resistance:mechanisms and pathogen fitness. Nat. Rev. Microbiol. 3 (2005) 547–556 |
| [7] | C. Lass-Flörl. Triazole antifungal agents in invasive fungal infections. Drugs. 71 (2011) 2405–2419 |
| [8] | J. Bartroil, E. Turmo, M. Alguero, et al. New azole antifungals. 3. Synthesis and antifungal activity of 3-substituted-4(3H)-quinazolinones1,2. J. Med. Chem. 41 (1998) 1869–1882 |
| [9] | F. Fratev, E. Benfenati. D-QSAR and molecular mechanics study for the differences in the azole activity against yeastlike and filamentous fungi and their relation to P450DM inhibition. 1.3-substituted-4(3H)-quinazolinones. J. Chem. Inf. Model. 45 (2005) 634–644 |
| [10] | L. Yu, Z.N. Liu, X.F. Cao, et al. Design and synthesis of pyridine-substituted itraconazole analogues with improved antifungal activities, water solubility and bioavailability. Bioorg. Med. Chem. Lett. 21 (2011) 4779–4783 |
| [11] | X.F. Cao, W.J. Chu, Y.B. Cao, Y.S. Yang. Design and synthesis of novel antifungal triazole derivatives with good activity and water solubility. Chin. Chem. Lett. 24 (2013) 303–306 |
| [12] | X.F. Cao, Z.S. Sun, Y.B. Cao, et al. Design, synthesis, and structure-activity relationship studies of novel fused heterocycles-linked triazoles with good activity and water solubility. J. Med. Chem. 57 (2014) 3687–3706 |
| [13] | W.Y. Wang, S.Z. Wang, Y. Liu, et al. Novel conformationally restricted triazole derivatives with potent antifungal activity. Eur. J. Med. Chem. 45 (2010) 6020–6026 |
| [14] | Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi. 2nd ed.; Approved Standard M38-A2; Clinical and Laboratory Standards Institute/National Committee for Clinical Laboratory Standards:Wayne, PA, 2008. |
| [15] | Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeast. 3rd ed.; Approved Standard M27-A3; Clinical and Laboratory Standards Institute/National Committee for Clinical Laboratory Standards:Wayne, PA, 2008. |
| [16] | D. Srikanta, F.H. Santiago-Tirado, T.L. Doering. Cryptococcus neoformans:historical curiosity to modern pathogen. Yeast. 31 (2014) 47–60 |
| [17] | X.F. Yu, V. Cojocaru, G. Mustafa, et al. Dynamics of CYP51:implications for function and inhibitor design. J. Mol. Recognit. 28 (2015) 59–73 |
| [18] | B.C. Monk, T.M. Tomasiak, M.V. Keniya. Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc. Natl. Acad. Sci. U S A. 111 (2014) 3865–3870 |