 2016, Vol. 27
2016, Vol. 27 


 , Muhammad Alib, Shifa Ullaha,c, Umer Rashida,b, Hayat Ullaha, Muhammad Tahad,e, Muhammad Tariq Javeda, Wajid Rehmana, Aftab Ahmad Khana, Obaid Ur Rahman Abida, Muhammad Bilalf
, Muhammad Alib, Shifa Ullaha,c, Umer Rashida,b, Hayat Ullaha, Muhammad Tahad,e, Muhammad Tariq Javeda, Wajid Rehmana, Aftab Ahmad Khana, Obaid Ur Rahman Abida, Muhammad Bilalf
b Department of Chemistry, COMSATS Institute of Information Technology, Abbottabad 22060, KPK, Pakistan ;
c Department of Biochemistry and Biotechnology, The Islamia University of Bahawalpur, Bahawalpur 63100, Pakistan ;
d Atta-ur-Rahman Institute for Natural Product Discovery, Universiti Teknologi MARA(UiTM), PuncakAlam Campus, 42300 Bandar PuncakAlam, Selangor DarulEhsan, Malaysia ;
e Faculty of Applied Science, UiTM, 40450 Shah Alam, Selangor, Malaysia ;
f Department of Environmental Science, COMSATS Institute of Information Technology, Abbottabad 22060, KPK, Pakistan
Urease (EC 3.5.1.5,urea amidohydrolase) is a nickel-dependent metalloenzyme that catalyzes the hydrolysis of urea to ammonia and CO2 or carbamate [1]. A variety of ureases are found in bacteria,fungi,higher plants and soil as a soil enzyme [2]. Activity of urease has been shown to be a prominent virulence determinant in the pathogenesis of many clinical conditions,which are detrimental for human and animal health as well as for agriculture [3]. Urease is known to be one of the major causes of diseases induced by Helicobacter pylori,thus allow them to survive at low pH inside the stomach. It also plays an important role in the pathogenesis of gastric and peptic ulcer [4]. Urease is directly involved during the formation of infectious stones and contributes to the pathogenesis of urolithiasis,pyelonephritis and hepatic encephalopathy,hepatic coma and urinary catheter encrustation [5]. Urease is responsible for urinary tract and gastrointestinal infections [6],possibly causing severe diseases such as peptic ulcers and stomach cancer as in the case of H. pylori [7]. In agriculture,high urease activity causes significant environmental and economic problems through releasing abnormally large amounts of ammonia into the atmosphere during urea fertilization [3]. Moreover,it induces plant damage primarily by depriving plants of their essential nutrients and secondarily by ammonia toxicity,increasing the pH of the soil [8]. Due to the diverse functions of this enzyme,its inhibition by potent and specific compounds could provide an invaluable addition for the treatment of infections and secondary complexes such as gastritis and gastric ulcer caused by H. pylori [9, 10].
Barbiturates are well-known compounds of hypnotic properties and are used as an active moiety on central nervous system. Thiobarbituric acid is different from barbituric acid due to the presence of a sulfur atom instead of an oxygen atom. Two active methylene hydrogen atoms at carbon-5 flanked between the two carbonyl carbons due to which their acidity further increased and their derivatives show significance biological activities [11]. Thiobarbituric acid analogs have been also reported as antifungal [12],antiurease [13],antimicrobial [14],Adiponectin expression [15],herbicides [16],anti-convulsing [17],anti-sclerosis’s agents [18] and antidiabetic and antibacterial agents [19]. Thiobarbituric acid analogs also showed anti-cancer and anti-viral activities [20].
2. Experimental1H NMR spectra were performed in DMSO-d6 on an Avance Bruker AM 300-500 MHz Instrument and TMS was used as an external reference. Chemical shifts values are given in δ (ppm). Electron impact mass spectra (EI-MS) were characterized on a Finnigan MAT-311A,Germany. Thin layer chromatography (TLC) was performed on pre-coated silica gel aluminum plates (Kieselgel 60,254,E. Merck,Germany). Chromatograms were visualized by UV at 254 and 365 nm.
2.1. General procedure for synthesis of bis-thiobarbiturate derivativesBis-thiobarbiturate derivatives 1-15 have been synthesized by the reactions of 1,3 diethyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (N,N-diethylthiobarbituric acid,2 mmol) with different aromatic aldehydes (1 mmol) in the presence of 5-10 mL of EtOH (Scheme 1). Reaction mixture was stirred in ethanol for about 3 h. The reaction completion was monitored by periodic TLC analysis. After the completion of the reactions,the mixture was poured over crushed ice followed by acidification with dill. HCl. Solid precipitates were collected by filtration,dried and recrystallized from ethanol to give the pure product in excellent yield. The structures of all synthetic compounds 1-15 were confirmed through EI-MS and 1H NMR.

|
Download:
|
| Scheme. 1. Synthetic protocol for bis-thiobarbiturate derivatives (1-15). | |
5,5'-((3,5-Dimethoxyphenyl)methylene)bis(1,3-diethyl-2- thioxodihydropyrimidine-4,6(1H,5H)-dione) (1): 1H NMR: (400 MHz,DMSO-d6): δ 7.2 (s,2H,H-2/6),6.8 (s,1H,H-4),3.7 (m,4H,CH2),3.5 (m,4H,CH2),3.4 (s,6H,OMe),3.1 (m,1H,CH),1.2 (m,12H,CH3); EI-MS: m/z (rel. int. %): 548 (M+,42),534 (45),350 (100),152 (34),99 (56).
5,5'-(Quinolin-2-ylmethylene)bis(1,3-diethyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione) (2): 1H NMR: (400 MHz,DMSO-d6): δ 7.8 (d,1H,J7,8 = 6.7 Hz,H-7),7.6 (d,1H,J8,7 = 6.7 Hz,H-8),7.3 (d,1H,J3,4 = 7.1 Hz,H-3),7.1 (d,1H,J6,5 = 7.5 Hz,H-6),6.8 (m,2H,H-4/ 5),3.6 (m,4H,CH2),3.4 (m,4H,CH2),3.0 (m,1H,CH),1.6 (m,12H,CH3); EI-MS: m/z (rel. int. %): 539 (M+,41),524 (75),351 (56),152 (100).
5,5'-((6-(Benzyloxy)-1H-indol-3-yl)methylene)bis(1,3-diethyl- 2-thioxodihydropyrimidine-4,6(1H,5H)-dione) (3): 1H NMR: (400 MHz,DMSO-d6): δ 7.8 (s,1H,H-2),7.6 (d,1H,J4,5 = 8.1 Hz,H-4),7.3 (d,1H,J5,4 = 7.5 Hz,H-5),7.1 (s,1H,H-7),6.8 (m,5H,H-2'/ 3'/4'/5'/6'),3.5 (m,4H,CH2),3.2 (m,4H,CH2),2.8 (m,1H,CH),1.4 (m,12H,CH3); EI-MS: m/z (rel. int. %): 633 (M+,56),514 (75),351 (100),162 (100).
5,5'-((3,4-Dihydroxyphenyl)methylene)bis(1,3-diethyl-2- thioxodihydropyrimidine-4,6(1H,5H)-dione) (4): 1H NMR: (400 MHz,DMSO-d6): δ 8.35 (s,1H,H-2),7.76 (dd,1H,J6,5 = 8.4,J6,2 = 2 Hz,H-6),7.0 (d,1H,J6,5 = 8.4 Hz,H-5),3.7 (m,4H,CH2),3.5 (m,4H,CH2),3.4 (s,6H,OMe),3.1 (m,1H,CH),1.2 (m,12H,CH3);EI-MS: m/z (rel. int. %): 520(M+,34),414(45),363(100),151 (37),99 (57).
2.2. Urease inhibition assayReaction mixtures having one unit of urease enzyme (Bacillus pasteurii) solution and 55 μL of buffers containing 100 mmol/L urea were incubated with 5 μL of test compounds (1 mmol/L) at 30 8C for 15 min in 96-well plates. Urease activity was determined by measuring the ammonia production using the indophenol’s method [21]. Briefly,45 μL of the phenol reagent and 70 μL of the alkali reagent were added to each well. The increased absorbance at 630 nm was measured after 50 min,using a micro-plate reader (Molecular Devices,USA). All reactions were performed in triplicate in a final volume of 200 μL. The results (change in absorbance per min) were processed using the Soft-Max Pro s4.5.Software (Molecular Devices,USA).
3. Results and discussion 3.1. ChemistryA set of 15 bis-thiobarbiturate derivatives 1-15 were synthesized by the reactions of N,N-diethylthiobarbituric acid with different aromatic aldehydes in ethanol as a solvent. The structures of all the synthesized compounds 1-15 were confirmed on the basis of spectral data. 1H NMR data of new bis-thiobarbiturate analogues were recorded and several generalizations could be made. Observation of a multiplet at 3.1 ppm is attributed to the bridged CH proton and is a clear indication of the product formation. A deshielded singlet with a two-proton integration appeared at 7.2 ppm,which could be assigned to the aromatic protons (H-2/6). The molecular ion peak was observed in the mass spectra of all the synthesized compounds,which confirmed their molecular masses.
3.2. Urease inhibition studiesBis-thiobarbiturate derivatives 1-15 were screened against the urease enzyme according to the literature protocol [21]. All compounds showed various degree of urease inhibitory activity,showing IC50 values ranging between 7.45 ± 0.12 and 74.24 ± 0.81 μmol/L,using thiourea as a standard (IC50 = 21 ± 0.11 μmol/L) (Table 1). Compounds 1,9 and 13 showed improved urease inhibitory activity as compared to the standard. The compounds 2-8,10-12 and 15 also showed good to moderate inhibitory activity against urease. However,compound 14 exhibited a weak inhibitory activity.
|
|
Table 1 Result of urease activity of bis-thiobarbiturate derivatives 1-15. |
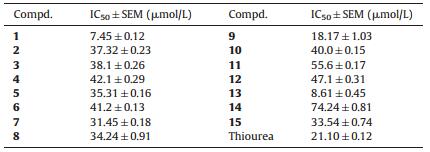
Structure activity relationship can be studied based on these data. The thiobarbituric acid remains constant and the only changes were the aromatic aldehydes. The SAR is mainly based on changes in the substitution pattern of the aromatic aldehydes. The 3,5- dimethoxy analog 1,N,N-dimethyl amino substituted analog 13 and meta-hydroxy analog 9 showed potent inhibitory activity in the series with IC50 values of 7.45 ± 0.12,8.61 ± 0.45 and 18.17 ± 1.03 μmol/L,respectively. The compounds 1 and 13 have electron donating groups on the phenyl ring. The activity might be due to these EDGs on the phenyl ring. In the case of compound 9,the hydroxy group on the phenyl ring might be responsible for this inhibition. The hydroxy might be involved in hydrogen bonding interactions with the nickel in the urease enzyme. Other hydroxy analogs such as 3,4-dihydroxy analog 4,2-naphthol substituted analog 7,p-hydroxy analog 8 and 2,3,4-trihydroxy analog 12,3-hydroxy-4-methoxy analog 11,and 3,5- tert-butyl-4-hydroxy analog 14 showed IC50 values of 42.1 ± 0.29,31.45 ± 0.18,34.24 ± 0.91,47.1 ± 0.31,55.6 ± 0.17 and 74.24 ± 0.81 μmol/L,respectively. The mono hydroxy-substituted analogs are found to possess better inhibitory activity than di- or tri-hydroxyl-substituted analogs. The position of the hydroxy group on the phenyl ring and other substituent also affects the activity. The compounds benzyloxy analog 10 and para-methoxy analog 15 showed good inhibition with IC50 values of 40.0 ± 0.15 and 33.54 ± 0.74 μmol/ L,respectively. Inhibitory activity of these compounds is less than that of compound 3,5-dimethoxy analog 1,although the EDG are present in the phenyl. The reason might be fewer substituents.
The quinoline-substituted analog 2 and indolyl analog 3 displayed IC50 values of 37.32 ± 0.23 and 53.12 ± 0.22 μmol/L,respectively. Both compounds have extra nitrogen atoms within the basic skeleton,and the activity might be due to those nitrogen atoms within the scaffold. The slight activity difference might be due to steric hindrance. The anthracenyl analog 5 and biphenyl analog 6 having IC50 values of 35.31 ± 0.16 and 41.2 ± 0.13 μmol/L also showed good inhibitory activity. The EDG might be responsible for this inhibition. In order to find the binding interactions of the most active analogs,molecular docking studies were performed.
3.3. Molecular docking studiesMolecular docking has contributed significantly in the identification of novel small drug-like scaffolds exhibiting high binding affinity and selectivity for the target of interest. Hence,we extended our study to investigate in silico binding orientation of the synthesized derivatives. The crystal structure of urease enzyme from B. pasteurii (PDB ID 4UBP) was selected for these studies. Docking experiments were performed using the Molecular Operating Environment (MOE) docking program [22]. The active site of urease is located within the cavity or the crevice in its internal territory in which an HAE molecule chelates with two nickel ions (Ni798 and Ni799) via hydroxyl oxygen. The key amino acid residues in the catalytic site of BPU are Ala170,His137,His139,Lys220,His249,His275,Gly280,Cys322,His323,His324,Arg339,Ala363 and Asp363. His137,His139 and KCX220,which interact with Ni799. Whereas,the His249,His275,Asp363 residues interact with Ni798. Carbamylated Lys490 (KCX220,a nonstandard residue),acts as a bridging residue between the two Ni ions.
We analyzed the computer generated molecular models of the compounds,and our analysis identified that all active compounds interact with the Ni ions in the urease enzyme. The coordination pattern of the most active compound 1 (IC50 = 7.45 ± 0.12 μmol/L) is shown in Fig. 1 (as viewed in Chimera 1.8.1 [22]). Compound 1 anchors itself in a way that enables a stronger coordination with the bi-nickel center (2.2 and 2.3 Å ,respectively) via its oxygen in the methoxy group at the 3-postion of the phenyl ring (Fig. 1).

|
Download:
|
| Fig. 1. 3D Modeled mode of binding of compound 1 into the active site of 4UBP. | |
To get better understanding of the roles of substituents on the aromatic ring,a docking analysis of compounds 13 and 9 was also carried out. According to the docked pose,N,N-dimethyl amino substituted analog 13 is well accommodated into the catalytic cavity,which allows the cyclic thiourea moiety of thio-barbiturate to coordinate tightly with the bi-nickel center (2.6 and 3.0 Å ,respectively,Fig. 2b). Similarly,compound 9 also showed a similar type of interactions to that of compound 13 (Fig. 2a). However,the distance between the thiourea moiety of thio-barbiturate and binickel center is 2.1 and 2.5 Å ,respectively. To rationalize the high urease inhibitory activity of compound 13 over 9,binding affinity of both compounds was analyzed. Compound 13 exhibited a docking score of -13.6152,a strong binding affinity of -11.403 kcal/mol and a low binding energy of -43.137 kcal/ mol. On the other hand,compound 9 was found to have a docking score of -12.9993,a binding affinity of -10.732 kcal/mol and a binding energy of -42.369 kcal/mol,which correlates with the in vitro data.

|
Download:
|
| Fig. 2. mode of binding of compounds 13 (a) and 9 (b) into the active site of urease from B. pasteurii (PDB ID 4UBP). | |
To further rationalize the comparison of molecular docking and in vitro results,docked pose of compound 14 was also investigated. Fig. 3 revealed that the presence of 3,5-di-tert-butyl-4-hydroxyphenyl group has a marked effect on potency. In compound 14,thio-barbiturate moiety displaced away from Ni798 (3.5 Å ) and showed no metal ligation,and this is a possible explanation for the poor in vitro activity of compound 14. Furthermore,compound 14 ranked lower having a docking score of -8.1966,a binding affinity of -6.591 kcal/mol and a binding energy of -29.861 kcal/mol.

|
Download:
|
| Fig. 3. (a) and (b) 3D and 2D binding interactions showing interaction of compound 14 into the binding site of 4UBP. | |
4. Conclusion
This study showed the bioorganic and medicinal chemists that simple one-step chemistry could generate extraordinary,bioactive compounds. During this study,we have synthesized 15 simple thiobarbituric acid derivatives,and evaluated their inhibitory activity against the urease enzyme. All compounds were identified as excellent urease inhibitors. This study discovered a novel class of urease inhibitors. The proposed scaffold of urease inhibitors offers a chance for further modifications that could give rise to lead structures with improved inhibitory activity and selectivity toward the enzyme.
| [1] | B. Krajewska, W. Zaborska. Double mode of inhibition-inducing interactions of 1,4-naphthoquinone with urease:Arylation versus oxidation of enzyme thiols. Bioorg. Med. Chem. 15 (2007) 4144–4151 |
| [2] | H.L.T. Mobley, R.P. Hausinger. Microbial ureases:significance, regulation, and molecular characterization. Microbiol. Rev. 53 (1989) 85–108 |
| [3] | Z. Amtul, N. Kausar, Atta-ur-Rahman, et al. Cysteine based novel noncompetitive inhibitors of urease(s)-Distinctive inhibition susceptibility of microbial and plant ureases. Bioorg. Med. Chem. 14 (2006) 6737–6744 |
| [4] | P.E. Wilcox. Chymotrypsinogens chymotrypsins. Methods Enzymol. 19 (1970) 64–108 |
| [5] | E. Bayerdörffer, R. Ottenjann. The role of antibiotics in Campylobacter pylori associated peptic ulcer disease. J. Gastroenterol. 23 (1988) 93–100 |
| [6] | C.M. Collins, S.E.F. D'Orazio. Bacterial ureases:structure, regulation of expression and role in pathogenesis. Mol. Microbiol. 9 (1993) 907–913 |
| [7] | C. Montecucco, R. Rappuoli. living dangerously:how Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell Biol. 6 (2001) 457–467 |
| [8] | G. Seneviratne, L.H.J. Van Holm, E.M.H.G.S. Ekanayake. Agronomic benefits of rhizobial inoculant use over nitrogen fertilizer application in tropical soybean. Field Crops. Res. 68 (2000) 199–203 |
| [9] | K.S.T. Ramsay, P. Wafo, Z. Ali, et al. Chemical constituents of Stereospermum acuminatissimum and their urease and a-chymotrypsin inhibitions. Fitoterapia 83 (2012) 204–208 |
| [10] | D.C. Menezes, E. Borges, M.F. Torres, J.P. Braga. A kinetic study of jack-bean urease denaturation by a new dithiocarbamate bismuth compound. Chem. Phys. Lett. 548 (2012) 85–89 |
| [11] | F. Zuccarello, B. Giuseppe, G. Concetta, C. Annalinda. Barbituric and thiobarbituric acids:a conformational and spectroscopic study. Spectrochim. Acta Part A:Mol. Biomol. Spectrosc. 59 (2003) 139–151 |
| [12] | M. Kidwai, R. Thakur, R. Mohan. Ecofriendly synthesis of novel antifungal (thio)-barbituric acid derivatives. Acta Chim. Solv. 52 (2005) 88–92 |
| [13] | K.M. Khan, F. Rahim, A. Khan, et al. Synthesis and structure-activity relationship of thiobarbituric acid derivatives as potent inhibitors of urease. Bioorg. Med. Chem. 22 (2014) 4119–4123 |
| [14] | L.K. Akopyan, A.S. Adzhibekyan, G.A. Porkinyan. Estimation of transition state and synthesis of barbituric acid with their derivatives of 1, 3, 4-thiadiazole. A. Etumasyan, Bilzh. Arm. 29 (1976) 80–83 |
| [15] | L. Ma, S. Li, H. Zheng, et al. Synthesis and biological activity of novel barbituric and thiobarbituric acid derivatives against non-alcoholic fatty liver disease. Eur. J. Med. Chem. 46 (2011) 2003–2010 |
| [16] | E.W. Brouwer, G.E. Felauerand, A.R. Bell. Synthesis, structure and solvatochromic properties of 3-cyano-4,6-diphenyl-5-(3- and 4-substituted phenylazo)-2-pyridones. U.S. Patent 779 (1990) 982 |
| [17] | S.V.K. Archana, A. Kumar. Synthesis of some newer derivatives of substitute quinazolinonyl-2-oxo/thiobarbituric acid as potent anticonvulsant agents. Bioorg. Med. Chem. 12 (2004) 1257–1264 |
| [18] | K.M. Khan, M. Ali, T.A. Farooqui, et al. An improved method for the synthesis of 5-arylidene barbiturates using BiCl3. J. Chem. Soc. Pak. 31 (2009) 823–828 |
| [19] | M. Hassan, K.A. Khan Faidallah. Synthesis and biological evaluation of new barbituric and thiobarbituric acid fluoro analogs of benzenesulfonamides as antidiabetic and antibacterial agents. J. Fluorine Chem. 142 (2012) 96–104 |
| [20] | D.T. Puerta, S.M.A. Cohen. A bioinorganic perspective on matrix metalloproteinase inhibition. Curt. Topic. Med. Chem. 4 (2004) 1551–1573 |
| [21] | M.W. Weatherburn. Phenol-hypochlorite reaction for determination of ammonia. Analyt. Chem. 39 (1967) 971–974 |
| [22] | Molecular Operating Environment (MOE), 2012.10; Chemical Computing Group Inc., Montreal, QC, Canada, H3A 2R7, 2012. |