 2016, Vol.27
2016, Vol.27 



b Collaborative Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin 300071, China
Infrared photodissociation (IRPD) spectroscopy can provide a wealth of structural information of gas-phase ions [1, 2, 3, 4]. For example, amino acids are known to be in their zwitterionic forms in solution and in their non-zwitterionic forms in isolated states. So interactions with other molecules or ions can greatly stabilize the zwitterionic structures of amino acids. In order to accurately deduce their gas-phase structures, a lot of amino acid relative complex ions were studied by combining the method of IRPD spectroscopy and theoretical calculations [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16]. Typically, these complex ions were generated by electrospray ionization (ESI) method, trapped in the cell of a Fourier transform ion cyclotron (FT ICR) mass spectrometer, and irradiated by a trunable IR laser. Then the spectral intensity of the selected ions at each wavelength can be calculated as:

Besides metal ions, amino acids themselves can greatly affect the structure of its partner in the corresponding cluster ions. For example, structures of several protonated homodimers of amino acids, including Gly2H+ [1, 5], Ser2H+ [1, 6], Thr2H+ [1, 16], Pro2H+ [5] and Lys2H+ [9, 15], have been studied by experimental IRPD method and theoretical calculations. IRPD spectrometry has been also applied for hetrodimers of amino acids and some magic clusters with larger size [6, 10, 13]. On the other hand, the chiral composition of amino acid clusters is also a very interesting and important topic, due to the basic question about the origins of chirality in biological systems. For example, the magic cluster of serine octamer is strongly characterized by its preference for homochirality [17, 18, 19, 20]. Research also showed that this cluster could be generated under simulated prebiotic conditions, indicating that its possible role in the chemistry of homochirogenesis [19]. Proline cluster is another interesting example [21, 22, 23, 24]. It could form a remarkably stable cluster ion of Pro12H+, which underwent spontaneous chiral resolution in the gas phase [22]. An oscillation between strong preferences for homochiral and heterochiral structures as a function of cluster size was also discovered [23]. A better understanding of the structures of these clusters and their relatives is very important. As a complementary method to ion mobility (IMS) mass spectrometry [24], IRPD spectroscopy can be applied in these clusters to help to identify their structural characterizes, such as the location of the proton and the hydrogen bonding interactions.
In the present work, IRPD method has been applied for the cluster ions of ʟ-Pro4H+ . And its experimental result was compared with the theoretical predicted spectra of the suggested isomers.
2. ExperimentalAll experiments were performed on a 7.0 T FT ICR mass spectrometer (IonSpec, Varian Inc., Lake Forest, CA, USA) in the positive ion mode. A solution of ʟ-Pro (1 mmol/L in 49:49:2 H2O:MeOH:AcOH) was sprayed at an infusion rate of 240 mL/h. A Zspray ESI source was used here. And the probe was set to 3.6 kV. After their generation, all ions were injected into an open-ended cylindrical Penning trap via an rf-only quadrupole ion guide. Then the precursor ions of Pro4H+ were further selected by the method of stored waveform inverse Fourier transform [25]. IRPD spectrum was obtained using the same experimental setup described previously [14, 15, 16]. The infrared OPO laser (Firefly-IR, M Squared, UK) was operated in the normal mode, with an output irradiation tunable from 2700 cm-1 to 4000 cm-1 and a line width of 7 cm-1. The average IR power was 400 mW. The irradiation time (with a typical value of 4 s) was controlled using a mechanical shutter (Sigma-Koki, Japan). The spectral intensity at each wavelength was calculated using (1).
For theoretical calculation, about 40 isomers of Pro4H+ were suggested based on the experimental IRPD spectra and previous studies [5, 24]. At the first step, these structures were optimized using the semi-empirical AM1 method. Then, 10 structures were selected, optimized, and verified by the DFT method of M062X/6- 31G(d) [26]. The four most stable isomers were further selected and optimized at the levels of M062X/6-31 + G(d, p). All frequencies obtained at the level of M062X/6-31 + G(d, p) were scaled with a factor of 0.948. The electronic energy was calculated at 0 K with zero-point energy corrections and free energies were calculated at 298 K. All calculations were carried out with the Gaussian 09 program [27]. The theoretical collision cross sections (ccs’s) of corresponding isomers were calculated with the MOBCAL program developed by Jarrold and coworkers [28, 29].
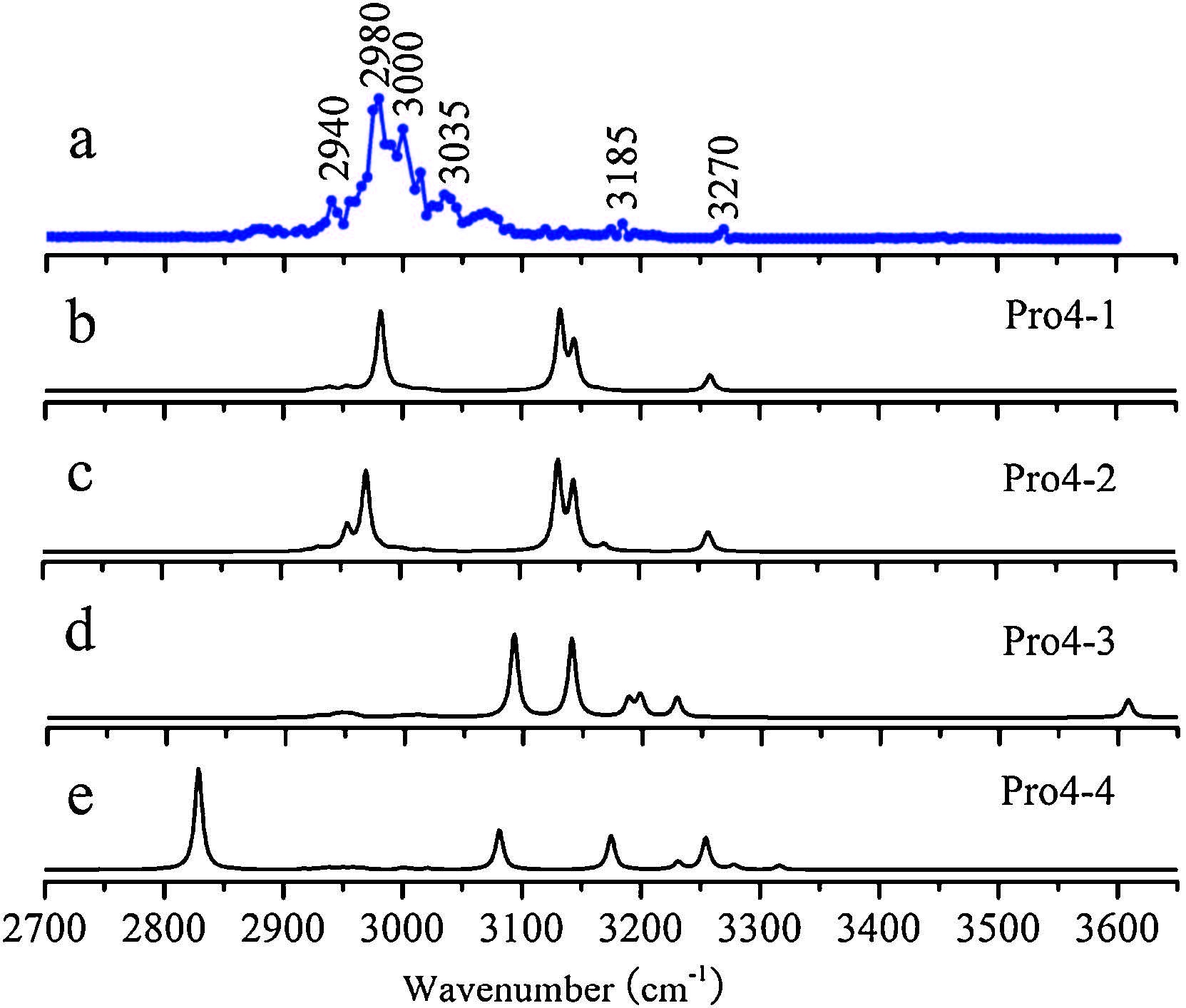
3. Results and discussionFig. 1a shows a typical ESI mass spectrum of proline. The cluster ions of Pro4H+ and Pro3H+ can be identified clearly. Weak signal of Pro3H+ can be observed too. For our purpose here, the ions of Pro4H+ were selected and isolated (Fig. 1b) and then irradiated by the IR laser. Under suitable wavelength, the absorbed IR photons can cause the dissociation of the cluster ions. Fig. 1c shows one example, at the wavenumbers of 2930 cm-1, the irradiation caused the parent ions dissociated to be cluster ions with smaller sizes (Pro3H+ and Pro2H+ observed here). By turning the wavelength of the IR laser and recording corresponding mass spectra, the IRPD spectrum can be calculated according to (1). Fig. 2a shows the IRPD spectrum of Pro4H+ in the range 2700-3600 cm-1 . A broad peak centered at 2980 cm-1 was observed. Some weak peaks at 2940 cm-1, 3035 cm-1, 3075 cm-1, 3185 cm-1 and 3270 cm-1 were also observed.

|
Download:
|
| Fig. 1.(a) ESI mass spectrum of the solution of ʟ-Pro, (b) the mass spectrum of the isolated ion of Pro4H+ and (c) the IRPD mass spectrum of Pro4H+ obtained by the irradiation of IR laser at 2930 cm-1 with a period of 4 s. | |

|
Download:
|
| Fig. 2.Comparison of (a) the experimental IRPD spectrum of Pro4H+ and predicted IR spectra of isomers of (b) Pro4-1, (c) Pro4-2, (d) Pro4-3, (e) Pro4-4. The calculations were performed at the M062X/6-31 + G(d, p) level and scaled with a single factor of 0.948. Structures of these isomers are shown in Fig. 3. | |
The structures of Pro4H+ were investigated using the calculation strategy described above. The most stable six isomers, which were optimized at the level of M062X/6-31 + G(d, p), are shown in Fig. 3. Their relative energies and free energies are summarized in Table 1. According to their total energy (at 0 K), the most stable structure is the isomer Pro4-1, which has an energy 0.34 kcal/mol lower than that of Pro4-2, and about 9-15 kcal/mol lower than those of other structures. However, Pro4-2 has the lowest value of Gibbs free energy (at 298 K), which is 0.19 kcal/mol lower than that of Pro4-1. Interestingly, all the top five conformations are shown to be arranged into geometry of a tetrahedral type. These structures are also very close to the previously suggested structure of D-Pro4H+ by Clemmer et al. [24]. Those isomers with geometry of a ring type are found to be higher in their energies. It is also found that the three unprotonated proline units in the top 5 structures are all characterized by their zwitterionic forms. For the most stable isomer Pro4-1, it has six strong intermolecular H-bonds of NH…O and one of OH…O, whose distances are all less than 1.90 Å . These H-bonds are greatly enhanced by the Coulomb interaction. The structure of isomer Pro4-2 is very close to the Pro4-1. It also has totally seven intermolecular H-bonds.

|
Download:
|
| Fig. 3.Six energetic minima of Pro4H+ optimized at the M062X/6-31 + G(d, p) level. | |
|
|
Table 1 Relative energies, free energies (both in kcal/mol) and theoretical ccs (in Å2) of different isomers of Pro4H+. |

{kind=link}
{kind=link}
{kind=link}
Theoretically predicted IR spectra of the four most stable isomers at M062X/6-31 + G(d, p) level were compared with the experimental IRPD spectrum and shown in Fig. 2. The predicted spectra of Pro4-1 (Fig. 2b) and Pro4-2 (Fig. 2c) are both characterized by strong absorptions at 2978 cm-1 (owing to the asymmetrical stretching mode of CH2), which are in good consistent with the observed peak at the 2980 cm-1. Both isomers predict peaks at ~3260 cm-1 that are also consistent with the experimental peak at 3270 cm-1. However, the predict peaks at ~3145 cm-1 (due to the NH2 symmetric stretching vibrational modes) are much stronger than the observed one. The reason is not very clear, but it is well known that the IRPD process is highly dynamic and possible isomerization process may cause the dissociation less efficient [4]. In contrast, the theoretical spectra of Pro4-3 and Pro4-4 agree with the experimental spectrum poorly.
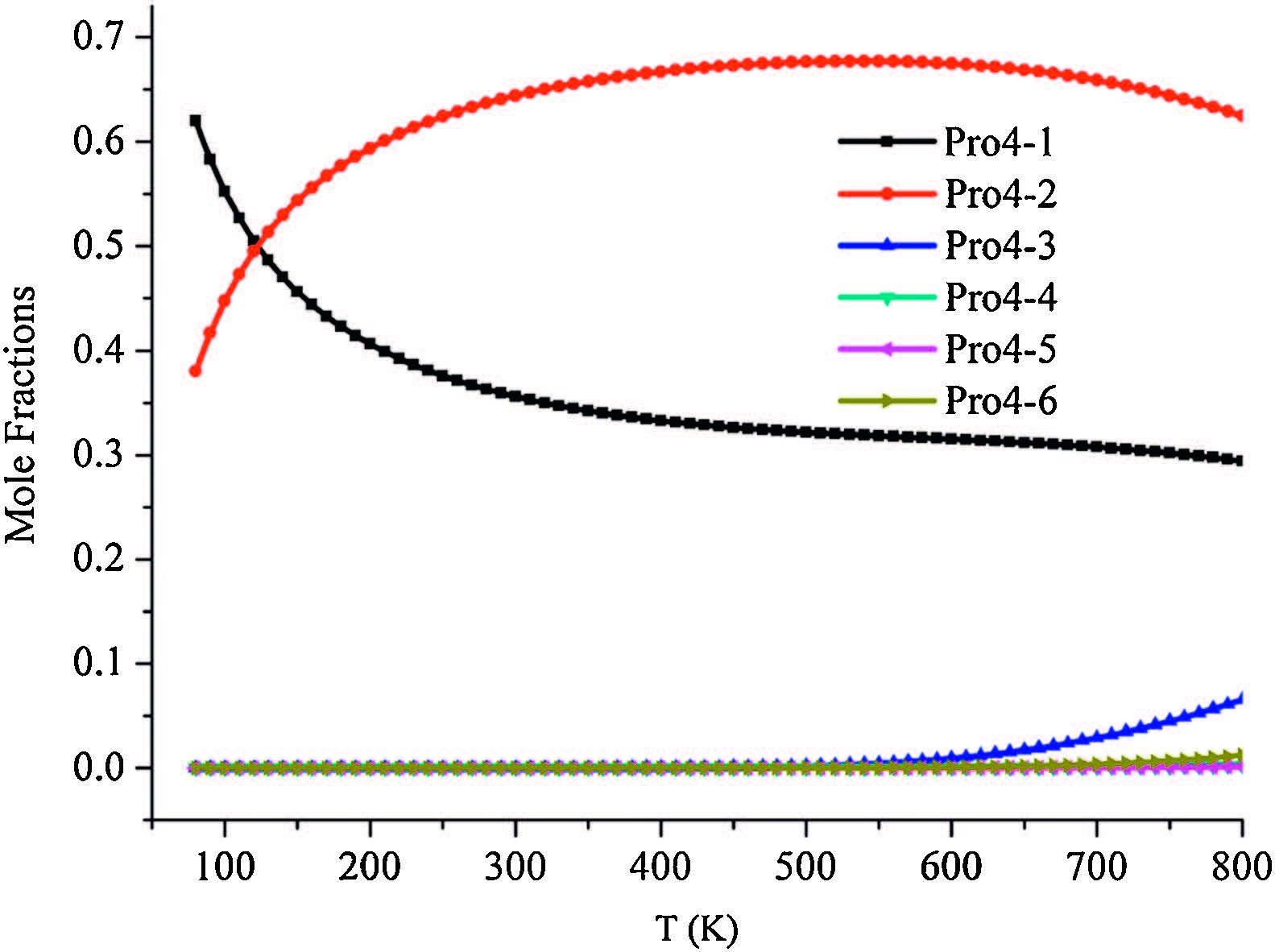
To further investigate the entropy effects andevaluate the relative concentrations of different isomers, mole fractions Wi of the six isomers proposed in Fig. 3 were also computed using the respective partition functions within the rigid-rotor and harmonic-oscillator approximation. The weight factor Wi of the ith isomer can be calculated according to:
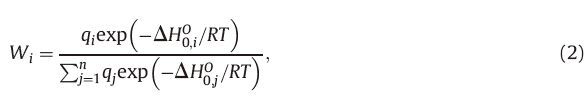

|
Download:
|
| Fig. 4.Temperature dependency of the relative concentrations of the six isomers of Pro4H+, based on calculations obtained on the level of M062X/6-31 + G(d, p). | |
{kind=link}
The ccs of a molecule is defined as the area around it in which the center of other molecule must be in order for a collision to occur. The value is sensitive to the three-dimensional structure of the molecule. The experimental ccs could be measured by the ion mobility mass spectrometry. For example, Clemmer et al. have experimentally measured the ccs of Pro4H+, which is 140.5 Å2 [24]. Here theoretical ccs’s of the six isomers are also calculated with the program of MOBCAL and shown in Table 1 [28, 29]. It can be found that the theoretical ccs of Pro4-1 is 140.8 Å2, which is very close to the experimental result. However, the ccs of Pro4-2 is found to be 142.8 Å2, which is 1.6% higher than the experimental one. Other isomers also have larger ccs’s than the experimental result.
4. ConclusionThe cluster ions of ʟ-Pro4H+ were generated by ESI method and studied using the FT ICR mass spectrometer. The experimental IRPD spectrum of Pro4H+ was obtained in the range of 2700-3600 cm-1 . A strong, broad peak centered at 2980 cm-1 was observed. Based on calculations at the M062X/6-31 + G(d, p) level, it was revealed that two most stable isomers, Pro4-1 and Pro4-2, had similar structures and energies. Their predicted IR spectra are also similar, and both of them are in good agreement with the experimental result. The calculated ccs of Pro4-1 is 140.8 Å2, which is very close to the reported experimental result of 140.5 Å2. These results also indicate that the units of proline should be characterized by their zwitterionic structures in the clusters.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (Nos. 21172121, 21475065) and the Fundamental Research Funds for the Central Universities is gratefully acknowledged.
| [1] | H.B. Oh, C. Lin, H.Y. Hwang, H. Zhai, et al., Infrared photodissociation spectroscopy of electrosprayed ions in a Fourier transform mass spectrometer, J. Am. Chem. Soc. 127(2005) 4076-4083. |
| [2] | J.R. Eyler, Infrared multiple photon dissociation spectroscopy of ions in penning traps, Mass Spectrom. Rev. 28(2009) 448-467. |
| [3] | N.C. Polfer, J. Oomens, Vibrational spectroscopy of bare and solvated ionic complexes of biological relevance, Mass Spectrom. Rev. 28(2009) 468-494. |
| [4] | N.C. Polfer, Infrared multiple photon dissociation spectroscopy of trapped ions, Chem. Soc. Rev. 40(2011) 2211-2221. |
| [5] | R. Wu, T.B. McMahon, Infrared multiple photon dissociation spectra of proline and glycine proton-bound homodimers. Evidence for zwitterionic structure, J. Am. Chem. Soc. 129(2007) 4864-4865. |
| [6] | X.L. Kong, I.A. Tsai, S. Sabu, et al., Progressive stabilization of zwitterionic structures in[H(Ser)2-8] + studied by infrared photodissociation spectroscopy, Angew. Chem. Int. Ed. 45(2006) 4130-4134. |
| [7] | M.F. Bush, J. Oomens, R.J. Saykally, E.R. Williams, Effects of alkaline earth metal ion complexation on amino acid zwitterion stability:results from infrared action spectroscopy, J. Am. Chem. Soc. 130(2008) 6463-6471. |
| [8] | R.C. Dunbar, J.D. Steill, J. Oomens, Cationized phenylalanine conformations characterized by IRMPD and computation for singly and doubly charged ions, Phys. Chem. Chem. Phys. 12(2010) 13383-13393. |
| [9] | R. Wu, R.A. Marta, J.K. Matens, K.R. Eldridge, T.B. McMahon, Experimental and theoretical investigation of the proton-bound dimer of lysine, J. Am. Soc. Mass. Spectrom. 22(2011) 1651-1659. |
| [10] | U.J. Lorenz, T.R. Rizzo, Multiple isomers and protonation sites of the phenylalanine/serine dimer, J. Am. Chem. Soc. 134(2012) 11053-11055. |
| [11] | Y.J. Alahmadi, A. Gholami, T.D. Fridgen, The protonated and sodiated dimers of proline studied by IRMPD spectroscopy in the N-H and O-H stretching region and computational methods, Phys. Chem. Chem. Phys. 16(2014) 26855-26863. |
| [12] | P.B. Armentrout, M.T. Rodgers, J. Oomens, J.D. Steill, Infrared multiphoton dissociation spectroscopy of cationized serine:effects of alkali-metal cation size on gas-phase conformation, J. Phys. Chem. A 112(2008) 2258-2267. |
| [13] | X.L. Kong, C. Lin, G. Infusini, et al., Numerous isomers of serine octamer ions characterized by infrared photodissociation spectroscopy, ChemPhysChem 10(2009) 2603-2606. |
| [14] | G.H. Liao, Y.J. Yang, X.L. Kong, Chirality effects on proline-substituted serine octamers revealed by infrared photodissociation spectroscopy, Phys. Chem. Chem. Phys. 16(2014) 1554-1562. |
| [15] | X.L. Kong, Reinvestigation of the structure of protonated lysine dimer, J. Am. Soc. Mass. Spectrom. 25(2014) 1-5. |
| [16] | H. Yin, X.L. Kong, Structure of protonated threonine dimers in the gas phase:saltbridged or charge-solvated, J. Am. Soc. Mass. Spectrom. 26(2015) 1455-1461. |
| [17] | R.G. Cooks, D. Zhang, K.J. Koch, F.C. Gozzo, M.N. Eberlin, Chiroselective selfdirected octamerization of serine:implications for homochirogenesis, Anal. Chem. 73(2001) 3646-3655. |
| [18] | Z. Takats, S.C. Nanita, R.G. Cooks, G. Schlosser, K. Vekey, Atmospheric pressure gas-phase H/D exchange of serine octamers, Anal. Chem. 75(2003) 6147-6154. |
| [19] | Z. Takats, S.C. Nanita, R.G. Cooks, Serine octamer reactions:indicators of prebiotic relevance, Angew. Chem. Int. Ed. 42(2003) 3521-3523. |
| [20] | S.C. Nanita, R.G. Cooks, Chiral enrichment of serine via formation, dissociation, and soft-landing of octameric cluster ions, Angew. Chem. Int. Ed. 45(2006) 554-559. |
| [21] | R.R. Julian, S. Myung, D.E. Clemmer, Do homochiral aggregates have an entropic advantage? J. Phys. Chem. B 109(2005) 440-444. |
| [22] | S. Myung, K.P. Lorton, S.I. Merenbloom, et al., Evidence for spontaneous resolution of icosahedral proline, J. Am. Chem. Soc. 128(2007) 15988-15989. |
| [23] | A.E. Holliday, N. Atlasevich, S. Myung, et al., Oscillations of chiral preference in proline clusters, J. Phys. Chem. A 117(2013) 1035-1041. |
| [24] | A.E. Holliday, N. Atlasevich, S.J. Valentine, D.E. Clemmer, Chirality and packing in small proline clusters, J. Phys. Chem. A 116(2012) 11442-11446. |
| [25] | R.B. Cody, R.E. Hein, S.D. Goodman, A.G. Marshall, Stored waveform inverse fourier transform excitation for obtaining increased parent ion selectivity in collisionally activated dissociation:preliminary results, Rapid Commun. Mass Spectrom. 1(1987) 99-102. |
| [26] | X. Zhao, D.G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements:two new functionals and systematic testing of four M06-class functionals and 12 other functional, Theor. Chem. Account. 120(2008) 215-241. |
| [27] | M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian09, Gaussian Inc, Wallingford, CT, 2009. |
| [28] | A.A. Shvartsburg, M.F. Jarrold, An exact hard-spheres scattering model for the mobilities of polyatomic ions, Chem. Phys. Lett. 261(1996) 86-91. |
| [29] | M.F. Mesleh, J.M. Hunter, A.A. Shvartsburg, G.C. Schatz, M.F. Jarrold, Structural information from ion mobility measurements:effects of the long-range potential, J. Phys. Chem. 100(1996) 16082-16086. |
| [30] | Z. Slanina, Equilibriumisomericmixtures:potential energy hypersurfaces as the origin of the overall thermodynamics and kinetics, Int. Rev. Phys. Chem. 6(1987) 251-267. |