 2016, Vol.27
2016, Vol.27 



b. Key Laboratory of Tropical Medicinal Plant Chemistry of Ministry of Education, Hainan Normal University, Haikou 571158, China;
c. State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China
After the pioneering work of Forrest, Thompson, and coworkers [1] over the past decade, many efforts have been devoted toward the design, synthesis, and photophysical investigation of new phosphorescent cyclometalated iridium(Ⅲ) complexes [2, 3, 4, 5, 6, 7, 8, 9, 10, 11]. Substantial research has demonstrated that the chemical structures of cyclometalated ligands (C^N ligands) can strongly affect the photophysical properties of iridium(Ⅲ) complexes. On the other hand, the introduction of a strongly electron-withdrawing group in the C^N ligand could cause a great increase of the luminescence efficiency [12, 13].
In pursuit of highly emissive iridium complexes, most recently, we have reported a series of cyclometalated phenylisoquinoline- based iridium(Ⅲ) complexes containing different electron-withdrawing substituents (-CF3, -CHO, -CN and -F) [14]. Their photophysical and electrochemical properties have been investigated, showing that the introduction of the electronwithdrawing fluorine group in the C^N ligand could improve phosphorescence quantum yields effectively. These results prompted us to study how the fluorine group in different positions of C^N ligands affect the pertinent emission properties. Hence, two new fluorine-modificative phenylisoquinoline iridium( Ⅲ) complexes (3a and 3b) have been prepared in this paper (Scheme 1). The spectroscopic and luminescence properties of these complexes are studied, and the lowest-energy electronic transitions are calculated with density functional theory (DFT) and time-dependent DFT (TD-DFT).

|
Download:
|
| Scheme 1.Synthetic routes of Ir(Ⅲ) complexes 3a-3b. | |
1H NMR spectra were recorded on a Bruker AM 400 MHz instrument. Chemical shifts were reported in ppm relative to Me4Si as the internal standard. ESI-MS spectra were recorded on an Esquire HCT-Agilent 1200 LC/MS spectrometer. The elemental analyses were performed on a Vario EL Cube Analyzer system. UV- vis spectra were recorded on a Hitachi U3900/3900H spectrophotometer. Fluorescence spectra were carried out on a Hitachi F-7000 spectrophotometer.
All air-sensitive and/or water-sensitive reactions were carried out under a dry nitrogen atmosphere. All commercial chemicals were used without further purification unless otherwise stated. Solvents were dried and degassed following standard procedures. Column chromatography was carried out using 200-300 μm mesh silica.
2.1. Preparation of 1-(2, 4-difluorophenyl)isoquinoline (f2piq, 1a)A mixture of 1-chloroisoquinoline (2.0 mmol), (2, 4-difluorophenyl) boronic acid (2.2 mmol), Pd(dppf)2Cl2 (5-10 mg) and potassium acetate (4.0 mmol) was dissolved in anhydrous 1, 4- dioxane (10 mL) and heated at 90 ℃ under nitrogen for 10 h. After cooling to room temperature, the mixture was poured into water (20 mL) and extracted with dichloromethane. The combined organic layers were dried over Na2SO4 and concentrated in vacuum. The crude product was purified by column chromatography to afford pure product 1a as a white solid. Yield: 93%. 1H NMR (400 MHz, CDCl3): δ 8.56 (d, 1H, J = 5.2 Hz), 7.83 (d, 1H, J = 8.0 Hz), 7.71 (d, 1H, J = 8.4 Hz), 7.64-7.66 (m, 2H), 7.46-7.51 (m, 2H), 7.00 (t, 1H, J = 8.4 Hz), 6.93 (t, 1H, J = 9.2 Hz). MS-ESI: m/z calculated: 241.1; found: 242.1 (M + 1), 264.1 (M + Na).
2.2. Preparation of 1-(4-fluoro-2-methylphenyl)isoquinoline (fmpiq, 1b)A procedure analogous to that for 1a was employed with (4- fluoro-2-methylphenyl)boronic acid instead of 2,4-difluorophenyl) boronic acid to afford 1b as a white solid. Yield: 90%. 1H NMR (400 MHz, CDCl3): δ 8.62 (d, 1H, J = 5.6 Hz), 7.89 (d, 1H, J = 8.0 Hz), 7.83 (d, 1H, J = 8.0 Hz), 7.68-7.72 (m, 2H), 7.54 (t, 1H, J = 8.0 Hz), 7.44 (t, 1H, J = 7.6 Hz), 7.13 (d, 1H, J = 8.0 Hz), 7.05 (d, 1H, J = 8.0 Hz), 2.47 (s, 3H). MS-ESI: m/z calculated: 237.1; found: 238.1 (M + 1), 260.1 (M + Na).
2.3. Preparation of [Ir(f2piq)2(bipy)][PF6] (3a)A mixture of IrCl3-3H2O (1.0 mmol) and the ligand 1a in 6 mL of ethoxyethanol and H2O (v/v = 2: 1) was refluxed for 12 h under nitrogen. Upon cooling to room temperature, the orange precipitate was collected by filtration and washed with cooled MeOH. After drying, the crude product of cholorobridged dimer complex 2a was used directly in next step without further purification. A mixture of the above dimer complex 2a and 2,2'-bipyridine (N^N ligand, 2.0 equiv.) was dissolved in 6 mL of DCM and MeOH (v/v = 2: 1) and heated at 40 ℃ under nitrogen for 6 h. After cooling to room temperature, NH4PF6 (2.5 equiv.) was added to the above solution. The mixture was stirred at room temperature for 3 h, and then evaporated to dryness. The solid was purified by column chromatography to afford pure product 3a, Yield: 63%. 1H NMR (400 MHz, CDCl3): δ 8.70 (d, 2H, J = 8.0 Hz), 8.27 (t, 2H, J = 9.2 Hz), 8.16 (t, 2H, J = 8.0 Hz), 7.88 (d, 2H, J = 8.0 Hz), 7.67-7.79 (m, 8H), 7.54 (t, 2H, J = 6.4 Hz), 7.46 (d, 2H, J = 6.0 Hz), 7.20 (d, 2H, J = 6.0 Hz), 6.61 (t, 2H, J = 9.2 Hz). MS-ESI: m/z calculated: 829.2; found: 829.3 (M-PF6). Anal. Calcd. for C40H24F10IrN4P: C, 49.33; H, 2.48, N, 5.75. Found: C, 49.56, H, 2.53, N, 5.91.
[Ir(fmpiq)2(bipy)][PF6] (3b) was obtained by a method similar to the preparation of 3a. Yield: 73%. 1H NMR (400 MHz, CDCl3): δ 8.74 (d, 2H, J = 7.2 Hz), 8.18 (t, 2H, J = 7.6 Hz), 8.16-8.20 (m, 3H), 8.16- 8.20 (m, 3H), 7.84-7.89 (m, 3H), 7.76 (t, 3H, J = 7.6 Hz), 7.66 (t, 3H, J = 7.6 Hz), 7.43-7.47 (m, 4H), 7.25-7.28 (m, 2H), 6.68 (t, 2H, J = 9.6 Hz), 2.31 (s, 6H). MS-ESI: m/z calculated: 821.2; found: 821.3 (M-PF6). Anal. Calcd. for C42H30F8IrN4P: C, 52.23; H, 3.13, N, 5.80. Found: C, 52.48, H, 3.21, N, 5.82.
2.4. Crystal structure determinationX-ray diffraction data were collected with an Agilent Technologies Gemini A Ultra diffractometer equipped with graphitemonochromated Mo Kα radiation (λ = 0.71073 Å) and Cu Kα radiation (λ = 1.5418 Å) at room temperature. Data collection and reduction were processed with CrysAlisPro software [15]. The structure was solved and refined using Full-matrix least-squares based on F2 with program SHELXS-97 and SHELXL-97 [16] within Olex2 [17]. During the refinement, the SQUEEZE option in the program PLATON was used to exclude the contribution from the highly disordered PF6 anion that cannot be modeled even with restraints [18, 19]. The 131 electrons voided by SQUEEZE closely corresponds to 1.87 PF6 anion (131 electrons). All non-hydrogen atoms were found in alternating difference Fourier syntheses and least-squares refinement cycles and, during the final cycles, refined anisotropically. Hydrogen atoms were placed in calculated positions and refined as riding atoms with a uniform value of Uiso. Crystallographic data (excluding structure factors) for the structural analyses have been deposited with the Cambridge Crystallographic Data Centre, Nos. 1062895 and 1062896 for 3a and 3b, respectively.
2.5. Computational detailsAll calculations were carried out with Gaussian 09 software package [20]. The density functional theory (DFT) and timedependent DFT (TD-DFT) were employed with no symmetry constraints to investigate the optimized geometries and electron configurations with the Becke three-parameter Lee-Yang-Parr (B3LYP) hybrid density functional theory [21, 22, 23]. The LANL2DZ basis set was used to treat the Ir atom, whereas the 6-31G* basis set was used to treat C, H, N and F atoms. Solvent effects were considered within the SCRF (self-consistent reaction field) theory using the polarized continuum model (PCM) approach to model the interaction with the solvent [24, 25].
2.6. Luminescence quantum efficiencyThe luminescence quantum efficiencies were calculated by comparison of the fluorescence intensities (integrated areas) of a standard sample fac-Ir(ppy)3 and the unknown sample according to the equation [26, 27, 28].
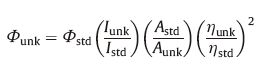
The crystals of 3a and 3b suitable for X-ray structural analysis were obtained by slow evaporation of CH2Cl2/MeOH solution. The crystallographic data and structure refinement details are given in Table 1; selected bond lengths and bond angles obtained by X-ray diffractions are collected in Table S1 in Supporting information.
|
|
Table 1 Crystallographic data and structure refinement details for complexes 3a, 3b. |
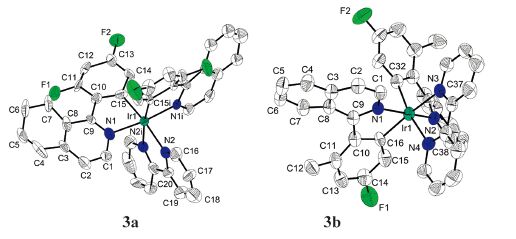
The perspective views of the cation of [Ir(f2piq)2(bipy)][PF6] and [Ir(fmpiq)2(bipy)][PF6] are depicted in Fig. 1. In the two similar crystal structures, iridium(Ⅲ) adopts a distorted [(C^N)2Ir(N^N)]+ octahedral geometry with the cis-C,C' and trans-N,N' configuration for phenylisoquinoline-based ligand. Simultaneously, the angles of atoms on the para positions of the octahedron range from 172.34(12)° to 173.61(15)° for 3a and 168.56(15)° to 171.91(17)° for 3b.

|
Download:
|
| Fig. 1.ORTEP views of 3a and 3b with the atom-numbering scheme at the 50% probability level. Hydrogen atoms and PF6 anion are omitted for clarity. | |
The Ir-C bonds of these complexes (Ir-Cav = 2.018 Å) are shorter than the Ir-N bonds (Ir-Nav = 2.099 Å). In particular, the Ir-N bond lengths from C^N ligands are significantly shorter than that from N^N ligands, consistent with strong trans influence of the carbon donors. All other bond lengths and angles within the chelate ligands are typical for this type of complexes [14, 29]. The bite angles of the phenylisoquinoline-based ligands (80.26° for 3a, 79.51° for 3b) and the bipyridine ligands (77.03° for 3a, 76.12° for 3b) lie in the same range as that observed in the literature for similar complexes [30]. In addition, the X-ray data show the large torsion angles (C8-C9-C10-C11, -31.358 for 3a and -31.308 for 3b), which suggest that the -F group can cause larger molecular deformation.
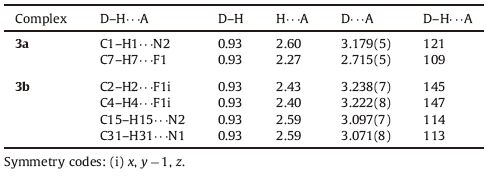
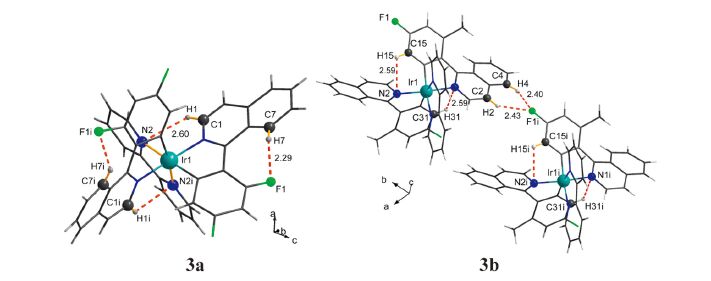
The hydrogen bond interactions in the crystal structures are presented in Fig. 2 and the details are summarized in Table 2. This conformation of 3a allows the establishment of two intramolecular C-H…F and C-H…N hydrogen bonds, making the sevenmembered and five-membered rings, respectively. However, the crystal structure of 3b exhibit both the intramolecular (C-H…N) and intermolecular (C-H…F) hydrogen bond interactions. And the molecular skeleton of 3b is linked by the C-H…F intermolecular hydrogen bonds to form a one-dimensional chain structure.

|
Download:
|
| Fig. 2.Part of the crystal structures of 3a and 3b, showing selected non-covalent contacts of the C-H…F and C-H…N types (dashed red lines). Atoms involved in hydrogen bonds are shown as balls of arbitrary radii. All other atoms and covalent bonds are represented as wires or sticks. Symmetry codes: (i)-x + 1, y, -z + 1/2 (3a); (i) x,y × 1, z (3b). | |
|
|
Table 2 Hydrogen bonding arrangements for 3a and 3b ( Å , °). |
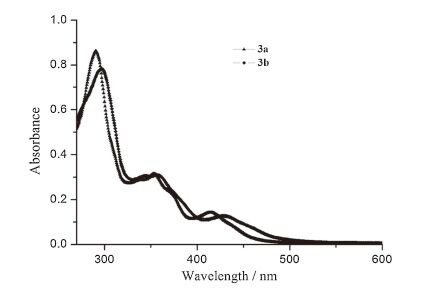
The UV-vis absorption spectra of 3a and 3b in CH2Cl2 solution at room temperature are presented in Fig. 3, and the data are provided in Table 3. All these complexes exhibit intense bands at the higher energies (250-350 nm), which are assigned to the spinallowed ligand-centered (LC) π-π* transitions. Somewhat weaker bands at lower energies (350-550 nm) are observed, which are attributed to both spin-allowed (singlet-to-singlet metal-to-ligand charge transfer, 1MLCT) and spin-forbidden (singlet-to-triplet metal-to-ligand charge transfer, 3MLCT) transitions [31, 32, 33, 34]. In comparison with 3a, the lowest lying absorption band for complex 3b is significantly red-shifted. The findings indicate that the substitution of F by CH3 could lower the HOMO-LUMO gap.

|
Download:
|
| Fig. 3.Electronic absorption spectra of 3a and 3b in CH2Cl2 at room temperature. | |
|
|
Table 3 Photophysical properties of complexes 3a and 3b. |
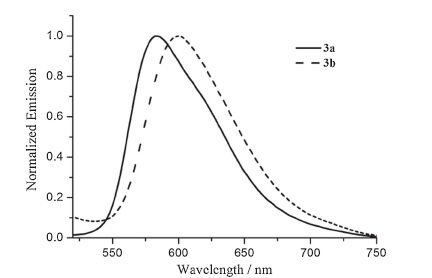
The normalized emission spectra of 3a and 3b in degassed CH2Cl2 solution at room temperature are given in Fig. 4, and the corresponding data are also listed in Table 3. The two iridium complexes exhibit broad and structureless emission bands centered at 584 and 600 nm, respectively, indicating that they are yellow and orange light-emitting materials. It is interesting to note the substituent changes in C^N ligand of 3b have a marked bathochromic effect on the phosphorescence spectra. It is also evident from Table S2 (Supporting information) that the substitution of F by CH3 on the C^N ligand increases the electron density around the iridium center (38.83→39.76%) and raises the highest occupied molecular orbital (HOMO) level (-6.083→-5.848 eV), thereby resulting in a reduced HOMO-LUMO energy gap (3.358→3.177 eV).

|
Download:
|
| Fig. 4.Normalized emission spectra of 3a and 3b in CH2Cl2 at room temperature. | |
Phosphorescence relative quantum yields (Φ) of 3a and 3b in dichloromethane solution were measured to be 0.074-0.11 (Table 3) at room temperature by using typical phosphorescent fac-Ir(ppy)3 as a standard (Φ = 0.40) [28]. The comparatively low phosphorescence yield observed for CH3-substituted complex 3b may be attributed to the intramolecular electron-transfer quenching of the excited state by the methyl group [35]. A comparison of the quantum yields observed for these complexes with the previously known related complex [Ir(C^N)2(bipy)]PF6 (C^N = 1- (3-fluoro-4-methylphenyl)isoquinoline) (Φ = 0.16) [14] is noteworthy. The results show the substitution at 3,4-positions is more effective than 2,4-positions.
3.4. Theoretical calculationsDensity functional theory (DFT) and time-dependent DFT (TDDFT) calculations were performed for complexes 3a and 3b to investigate the lowest-energy electronic transition of the ultraviolet absorption spectra. The most representative molecular frontier orbital diagrams are presented in Fig. 5. The calculated spin-allowed electronic transitions and electron density distributions are summarized in Table 4 and Table S2.

|
Download:
|
| Fig. 5.The frontier molecular orbital diagrams of complexes 3a and 3b from DFT calculations. | |
|
|
Table 4 Main experimental and calculated optical transitions for 3a and 3b. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
For all of the species investigated, the electron density of the highest occupiedmolecular orbital(HOMO) is centeredonthemetal with a substantial contribution from the cyclometalating ligands, as is also reported for similar iridium cyclometalated complexes [36, 37]. Conversely, the electron density in the lowest unoccupied molecular orbital (LUMO) locates mainly on the ancillary ligands. The successive unoccupied molecular orbital LUMO + 1 primarily originates fromthe cyclometalating ligands. The theory calculations of DFT reveal that the lowest-energy electronic transitions (438 nm for 3a, 459 nm for 3b) both arise from HOMO→LUMO + 1 orbital electronic transitions (Table 4). In comparison with 3a (3.583 eV), the reduction of energy gap for 3b (3.463 eV) is consistent with the red shift observed in absorption spectra.
4. ConclusionIn conclusion, two new fluorinated phenylisoquinoline-based iridium(Ⅲ) complexes [Ir(C^N)2(bipy)][PF6] (3a and 3b) have been synthesized and thoroughly characterized by studying their crystallographic, spectroscopic and luminescence properties. X-ray diffraction studies of complexes 3a and 3b reveal that they all possess pseudo-octahedral coordination geometry. The theoretical calculations have also been performed to rationalize the lowest-energy electronic transitions of ultraviolet absorption spectra. Photoluminescence is observed in the yellow (3a) and orange regions (3b) at room temperature with quantum yield of 0.074-0.11. Upon a change of substituent on cyclometalated ligand, the absorption and emission spectra exhibit a strong dependence on the chemical structures of the ligands. The results will facilitate the design of new phenylisoquinoline-based ligands for light-emitting iridium complexes.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 21501037), the Natural Science Foundation of Hainan Province (No. 20152017) and the Science and Research Project of Education Department of Hainan Province (Nos. Hjkj2013-25 and Hnky2015-27).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2015.12.007.
| [1] | M.A. Baldo, D.F. O'Brien, Y. You, et al., Highly emission from electroluminescent phosphorescent organic devices, Nature 395(1998) 151-154. |
| [2] | M.A. Baldo, S. Lamansky, P.E. Burrows, et al., Very high-efficiency green organic light-emitting devices based on electrophosphorescence, Appl. Phys. Lett. 75(1999) 4-6. |
| [3] | M.A. Baldo, C. Adachi, S.R. Forrest, et al., High-efficiency organic electrophosphorescent devices with tris(2-phenylpyridine)iridium doped into electrontransporting materials, Appl. Phys. Lett. 77(2000) 904-906. |
| [4] | S. Lamansky, P. Djurovich, D. Murphy, et al., Synthesis and characterization of phosphorescent cyclometalated iridium complexes, (Ⅰ)norg. Chem. 40(2001) 1704-1711. |
| [5] | N.G. Park, M.Y. Kwak, B.O. Kim, et al. Jpn. J. Appl. Phys. 41(2002) 1523-1526. |
| [6] | A.B. Tamayo, B.D. Alleyne, P.(Ⅰ). Djurovich, et al., Synthesis and characterization of facial and meridional tris-cyclometalated iridium(Ⅲ) complexes, J. Am. Chem. Soc. 125(2003) 7377-7387. |
| [7] | W.Y. Wong, C.L. Ho, Z.Q. Gao, et al., Multifunctional iridium complexes based on carbazole modules as highly efficient electrophosphores, Angew. Chem. (Ⅰ)nt. Ed. 45(2006) 7800-7803. |
| [8] | H.H. Chou, C.H. Cheng, A highly efficient universal bipolar host for blue, green, and red phosphorescent OLEDs, Adv. Mater. 22(2010) 2468-2471. |
| [9] | Y.T. Tao, Q.A. Wang, C.L. Yang, et al., Multifunctional triphenylamine/oxadiazole hybrid as host and exciton-blocking material:high efficiency green phosphorescent OLEDs using easily available and common materials, Adv. Funct. Mater. 20(2010) 2923-2929. |
| [10] | D. Sykes, (Ⅰ).S. Tidmarsh, A. Barbieri, et al., d→f energy transfer in a series of (Ⅰ)r(Ⅲ)/Eu(Ⅲ) dyads:energy-transfer mechanisms and white-light emission, (Ⅰ)norg. Chem. 50(2011) 11323-11339. |
| [11] | Y. Zheng, A.S. Batsanov, R.M. Edkins, et al., Thermally induced defluorination during a mer to fac transformation of a blue-green phosphorescent cyclometalated iridium(Ⅲ) complex, (Ⅰ)norg. Chem. 51(2012) 290-297. |
| [12] | C.L. Ho, W.Y. Wong, Q. Wang, et al., A multifunctional iridium-carbazolyl orange phosphor for high-performance two-element WOLED exploiting excitonmanaged fluorescence/phosphorescence, Adv. Funct. Mater. 18(2008) 928-937. |
| [13] | F. Babudri, G.M. Farinola, F. Naso, et al., Fluorinated materials for electronic and optoelectronic applications:the role of the fluorine atom, Chem. Commun. (2007) 1003-1022. |
| [14] | Z.G. Niu, D. Liu, J. Zuo, et al., Four new cyclometalated phenylisoquinoline-based (Ⅰ)r(Ⅲ) complexes:syntheses, structures, properties and DFT calculations, (Ⅰ)norg. Chem. Commun. 43(2014) 146-150. |
| [15] | Agilent Technologies (Ⅰ)nc., CrysAlisPro Version 1.171.36.21, Agilent Technologies (Ⅰ)nc., Santa Clara, CA, USA, 2012. |
| [16] | G.M. Sheldrick, A short history of shelx, Acta Crystallogr. Sect. A:Found. Crystallogr. 64(2008) 112-122. |
| [17] | O.V. Dolomanov, L.J. Bourhis, R.J. Gildea, et al., Olex2:a complete structure solution, refinement and analysis program, J. Appl. Cryst. 42(2009) 339-341. |
| [18] | A.L. Spek, Single-crystal structure validation with the program platon, J. Appl. Crystallogr. 36(2003) 7-13. |
| [19] | P.V.D. Sluis, A.L. Spek, Bypass:an effective method for the refinement of crystal structures containing disordered solvent regions, Acta Crystallogr. Sect. A:Found. Crystallogr. 46(1990) 194-201. |
| [20] | M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 09, Revision A. 01, Gaussian, (Ⅰ)nc, Wallingford, CT, 2009. |
| [21] | C. Lee, W. Yang, R.G. Parr, Development of the colle-salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B:Condens. Matter 37(1988) 785-789. |
| [22] | B. Miehlich, A. Savin, H. Stoll, et al., Results obtained with the correlation energy density functionals of becke and lee, yang and parr, Chem. Phys. Lett. 157(1989) 200-206. |
| [23] | A.D. Becke, Density-functional thermochemistry. Ⅲ. The role of exact exchange, J. Chem. Phys. 98(1993) 5648-5652. |
| [24] | M. Cossi, N. Rega, G. Scalmani, et al., Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem. 24(2003) 669-681. |
| [25] | J. Tomasi, B. Mennucci, R. Cammi, Quantum mechanical continuum solvation models, Chem. Rev. 105(2005) 2999-3093. |
| [26] | A. Juris, V. Balzani, F. Barigelletti, et al., Ru(Ⅱ) polypyridine complexes:photophysics, photochemistry, electrochemistry, and chemiluminescence, Coord. Chem. Rev. 84(1988) 85-227. |
| [27] | M. Frank, M. Nieger, F. Vögtle, et al., Dinuclear RuⅡ and/or OsⅡ complexes of bis, (Ⅰ)norg. Chim. Acta. 242(1996) 281-291. |
| [28] | K.A. King, P.J. Spellane, R.J. Watts, Excited-state properties of a triply orthometalated iridium(Ⅲ) complex, J. Am. Chem. Soc. 107(1985) 1431-1432. |
| [29] | S. Kammer, (Ⅰ). Starke, A. Pietrucha, et al., 1,12-diazaperylene and 2,11-dialkylated-1,12, Dalton Trans. 41(2012) 10219-10227. |
| [30] | M. Bandini, M. Bianchi, G. Valenti, et al., Electrochemiluminescent functionalizable cyclometalated thiophene-based iridium(Ⅲ) complexes, (Ⅰ)norg. Chem. 49(2010) 1439-1448. |
| [31] | S.K. Leung, K.Y. Kwok, K.Y. Zhang, et al., Design of luminescent biotinylation reagents derived from cyclometalated iridium(Ⅲ) and rhodium(Ⅲ) bis(pyridylbenzaldehyde) complexes, (Ⅰ)norg. Chem. 49(2010) 4984-4995. |
| [32] | T. Hofbeck, H. Yersin, The triplet state of fac-(Ⅰ)r(ppy)3, (Ⅰ)norg. Chem. 49(2010) 9290-9299. |
| [33] | Q.L. Xu, C.C. Wang, T.Y. Li, et al., Syntheses, photoluminescence, and electroluminescence of a series of iridium complexes with trifluoromethyl-substituted 2-phenylpyridine as the main ligands and tetraphenylimidodiphosphinate as the ancillary ligand, (Ⅰ)norg. Chem. 52(2013) 4916-4925. |
| [34] | M. Tavasli, T.N. Moore, Y.H. Zheng, et al., Colour tuning from green to red by substituent effects in phosphorescent tris-cyclometalated iridium(Ⅲ) complexes of carbazole-based ligands:synthetic, photophysical, computational and high efficiency OLED studies, J. Mater. Chem. 22(2012) 6419-6428. |
| [35] | K.R.J. Thomas, M. Velusamy, J.T. Lin, et al., Efficient red-emitting cyclometalated iridium(Ⅲ) complexes containing lepidine-based ligands, (Ⅰ)norg. Chem. 44(2005) 5677-5685. |
| [36] | M.S. Lowry, W.R. Hudson, R.A. Pascal Jr., et al., Accelerated luminophore discovery through combinatorial synthesis, J. Am. Chem. Soc. 126(2004) 14129-14135. |
| [37] | Q. Zhao, S.J. Liu, M. Shi, et al., Series of new cationic iridium(Ⅲ) complexes with tunable emission wavelength and excited state properties:structures, theoretical calculations, and photophysical and electrochemical properties, (Ⅰ)norg. Chem. 45(2006) 6152-6160. |