 2016, Vol.27
2016, Vol.27 



Lithium-ion batteries (LIBs) are among the most important rechargeable energy storage devices due to their wide range of applications, such as portable electronic devices and electrical/ hybrid vehicles. In order to meet the increasing demand for high power density in electrical/hybrid vehicles, much attention has been focused on many metal oxides with higher theoretical capacity than graphite [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15]. As one of the promising anode materials, GeO2 has a high theoretical capacity of 1126 mA h g-1 by forming Li4.4Ge alloy [1, 2, 3, 4], which is higher than other metal oxides, such as SnO2 [9, 10, 11, 14, 15], NiO [12], ZnO [7, 12], TiO2 [7], MnO [5], Fe2O3 [5], Co3O4 [8] and CuO [6]. However, like these metal oxides, GeO2 also exhibits poor electrical conductivity and significant volume expansion and contraction during the charging and discharging processes, which causes degradation of electrode cyclic performance [1, 2, 3, 4]. In such cases, other inactive or less active compositions are used as supporting and conducting matrix to buffer the volume change [12, 13, 14, 15]. Generally, carbon is considered one of the most effective matrices due to its excellent electrical conductivity and small volume variation during the processes of charging and discharging [1, 2, 13, 14, 15, 18]. At present, some GeO2-C composite anode materials for LIBs were usually synthesized by hydrothermal methods and subsequent coating processes [1, 2].
One-dimensional (1D) nanostructure electrode materials usually demonstrate excellent electrochemical performance because their unique 1D structure can provide a large contact area with electrolytes, shorten diffusion path for lithium ions and release mechanical strains between particles induced by the volume change during the processes of electrochemical reactions [10, 11, 12, 13, 14, 15]. The electrospinning is a relatively simple, inexpensive and large scale producing technique for preparing fibrous materials. Many metal oxides-carbon composite fibers can be fabricated by electrospinning solutions containing polymers, such as polyacrylonitrile (PAN), polyvinyl-pyrrol-idone (PVP) or polyvinyl-alcohol (PVA), and the precursors of metal oxides, and subsequently carbonizing the electrospun fibers in an inert atmosphere [13, 14, 15]. These metal oxides-carbon composite fibers possess high surface area, porous structure and electrical conductivity, which results from the oxidation of carbon during thermal process and interconnected morphologies between the interlaying metal oxides embedded carbon fibers [13, 14, 15].
However, to the best of our knowledge, the preparation of electrospun GeO2-C fibers and their application as high performance anode materials for LIBs have not been reported. In our present work, we synthesized GeO2-C fibers by electrospinning a spinnable sol containing PAN, PVP and GeO2 precursors followed by subsequent calcination in an inert atmosphere, and studied their electrochemical performance as anode materials of LIBs.
2. ExperimentalSynthesis of GeO2-C fibers: In a typical procedure of preparing spinnable sol, 0.5 g of GeCl4 was dropped into 7 g of N,N-dimethylformamide (DMF), and white precipitates formed immediately, and then 0.5 g of PAN and 0.5 g of PVP were added to form a homogeneous transparent sol by vigorous stirring until the white precipitates disappear completely. The sol was transferred into a 10 mL syringe with a 0.8 mm internal diameter needle. For electrospinning, the sol was pushed out of spinner at a feed speed of 0.3 mL h-1 with a high voltage of 12.5 kV and a distance of 12 cm between the spinner and collector. Finally, the electrospun fibers were stabilized at 280 ℃ for 3 h in air, and then carbonized at 600 ℃ for 2 h in Ar with a heating rate of 2 ℃ min-1.
The crystal structures were identified by X-ray diffraction (XRD: MMA, GBC) with Cu Kα radiation and a graphite monochromator. The morphologies observation and compositions analysis were carried out by field emission scanning electronmicroscopy (FESEM: JEOL 7500FA), transmission electronmicroscopy (TEM: JEOL 2011F) and energy dispersive X-ray spectroscopy (EDX: JEOL 2011F). X-Ray photoelectron spectroscopy (XPS) was conducted on a VG Scientific ESCALAB 220IXL instrument using aluminum Kα X-ray radiation during XPS analysis. Thermogravimetric analysis (TGA) was performed on aMETTLER TOLEDO TGA/DSC1 type instrument from 40 ℃ to 700 ℃ at a heating rate of 10 ℃ min-1 in air.
The working electrodes were prepared by mixing 85 wt% GeO2-C fibers and 15 wt% poly(vinyl difluride) (PVDF) in 1-methyl-2-pyrrolidinone (NMP). The mixture was ground until it became a homogeneous slurry and pasted on a piece of copper foil. The electrodeswere dried in a vacuumoven at 120 ℃ for 12 h to remove NMP. The electrochemical tests were carried out using CR2032 coin type cells, which were assembled in an argon-filled Mbraun glove box with pure lithium metal foil as a counter electrode. The electrolyte consists of a solution of 1 mol L-1 LiPF6 in a mixture of ethylene carbonate (EC), dimethyl carbonate (DMC) and diethyl carbonate (EC/DMC/DECwith a volume ratio of 3/4/3) with 5 wt% of fluoroethylene carbonate (FEC) additive. The cyclic performance was measured at a current density of 50 mA h g-1, and the rate performance was tested at different current densities of 50 mA h g-1, 200mA h g-1, 400mA h g-1, 600 mA h g-1, 800mA h g-1 and 50 mA h g-1, respectively, on a LAND-CT2001A instrument by the galvanostatic method. Cyclic voltammetry (CV) test was performed on an electrochemical workstation (CHI660A) at a scan rate of 0.1 mV s-1 between 0.01 V and 3.0 V.
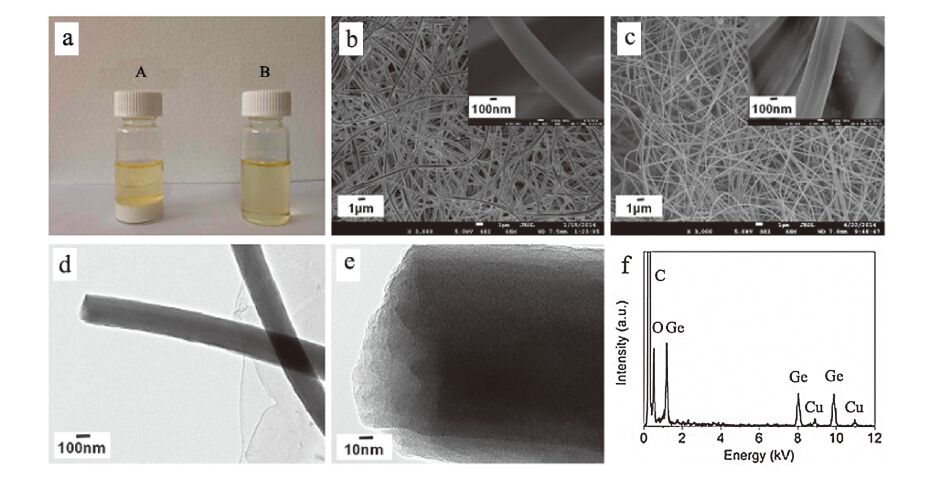
3. Results and discussionIn our experiment, the spinnable sol system was selected because GeCl4 react with some solvents, such as H2O, ethanol and DMF intensely, and form precipitates. Bottle A in Fig. 1a shows the white precipitates will not disappear even after a very long time of stirring by only adding PAN. If PVP was used to replace PAN, the precipitates will not disappear either. Bottle B in Fig. 1a indicates the PAN and PVP with a weight ratio of 1:1 can successfully disperse the precipitates and form a homogeneous transparent sol after vigorous stirring for several hours. Fig. 1b and its inset show the SEM images of the electrospun fibers. The fibers are uniform and have diameters of ~300 nm and lengths extending to several hundred microns. After being calcined in Ar at 600 ℃ for 2 h, their fibrous morphologies with almost the same diameters have been retained (Fig. 1c and its inset). TEM was employed to further observe and analyze their morphologies, structures and chemical compositions. Fig. 1d shows the fibers have diameters of about ~300 nm, which corresponds to the SEM images of Fig. 1c and its inset. The HRTEM image of calcined fibers in Fig. 1e displays that the fibers have no pores, indicating their dense structure. The selected-area electron diffraction (SEAD) pattern in inset of Fig. 1e reveals the fibers are amorphous. Fig. 1f shows the EDX pattern of the fiber in Fig. 1e. The fibers consist of Ge, O and C elements and the molar ratio of Ge and O is approximately 1:2, suggesting the calcined fibers perhaps are GeO2-C fibers. The peaks corresponding to Cu are contributed to the Cu grid used.

|
Download:
|
| Fig. 1. (a) digital picture of mixture with white precipitate by adding PAN (Bottle A) and sol by adding PAN and PVP with a weight ratio of 1:1 (Bottle B); (b) FESEMimages of electrospun fibers and a single fiber (inset); (c) FESEM images of calcined fibers and a single fiber (inset); (d) TEM image of calcined fibers; (e) HRTEM image of a calcined fiber and its SEAD patterns (inset); (f) EDX patterns of the calcined fiber in (e). | |
{kind=link}
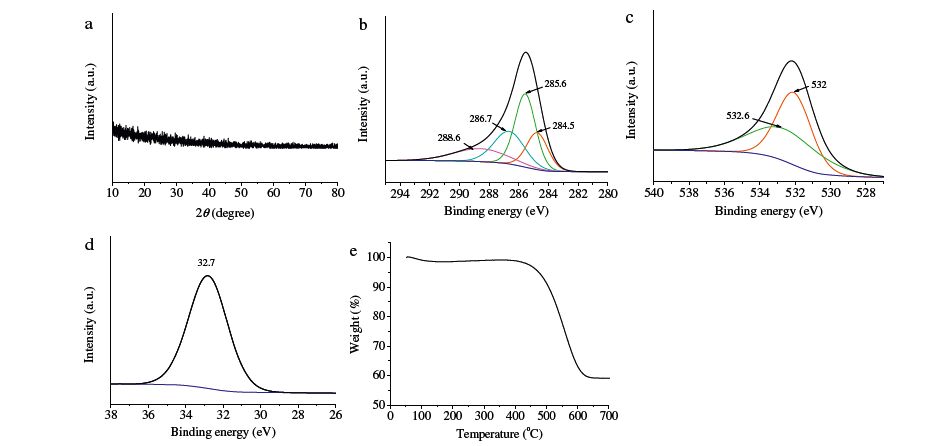
Fig. 2a shows the XRD pattern of GeO2-C fibers. No peaks can be observed in the XRD pattern, confirming their amorphous structure. XPS can provide much useful information about the elemental compositions of the compounds by analyzing the chemical state of their elements. Fig. 2b-d show the XPS spectra of the GeO2-C fibers. The observed peaks in Fig. 2b can be fitted into four overlapping peaks with bond energy of 284.5, 285.6, 286.7 and 288.6 eV, respectively. The peak at 284.5 eV is corresponding to graphitic carbon containing polyaromatic layered structures, the peak at 285.6 eV is attributed to disorder carbons, the peaks at 286.7 and 288.6 eV can be assigned to C=O groups [15, 16]. GeO2-C fibers have a particular one-dimensional structure with GeO2 encapsulated in the carbon matrix, so part of carbon will bond to GeO2 through the C=O group [15]. In Fig. 2c, the peak at 532 eV can be ascribed to the O2- ions in the GeO2 framework, while the other peak at 532.6 eV is attributed to the C=O group [18, 19]. In Fig. 2d, the peak at 32.7 eV is assigned to Ge4+ in the GeO2 [16, 17]. Therefore, the XPS results further confirm the GeO2-C fibers consist of GeO2 and amorphous carbon. TGA was used to measure their carbon content (Fig. 2e). Their weight loss can be divided into 3 stages: (1) when temperature rises from room temperature to 130 ℃, the weight loss was ~2 wt% due to evaporation of physically adsorbed H2O on the surface of the fibers; (2) from 130 ℃ to 400 ℃, their weight remained almost constant; (3) from 400 ℃ to 630 ℃, their weight decreased greatly, and the weight loss was about ~40 wt% because of the oxidation of carbon. Above 630 ℃, their weight did not change any more. So the GeO2-C composite fibers contain ~40 wt% carbon.

|
Download:
|
| Fig. 2.GeO2-C composite fibers: (a) XRD patterns; (b) XPS spectrum of C 1s; (c) XPS spectrum of O 1s; (d) XPS spectrum of Ge 3d5/2; (e) TGA curve. | |
{kind=link}
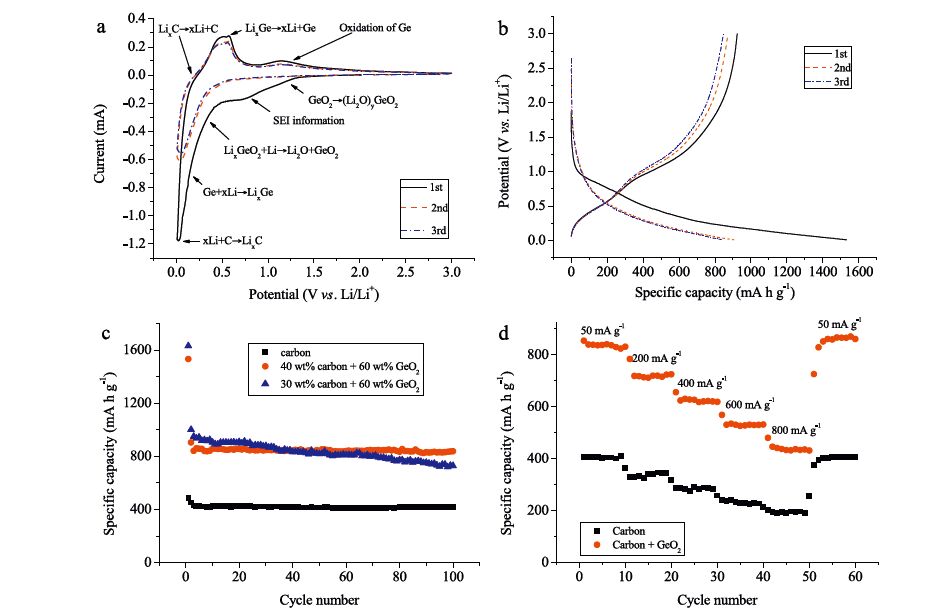
The electrochemical behavior of GeO2-C fibers was investigated by cycle voltammetry (CV) in a voltage range of 0.01-3.0 V (versus Li/Li+) at a scan rate of 0.1 mV s-1. Fig. 3a shows the CV curves of GeO2-C fiber electrode in the first, second and third scanning cycles. In the first cycle, the broad reduction peak from 0.5 V to 1.3 V is corresponding to multi-step electrochemical processes including the formation of an SEI layer, the formation of a vitreous phase lithium germanate and the subsequent reduction of GeO2 and LixGeO2 into Ge and Li2O [1, 2, 3, 4]. Because the formation of SEI layer in the first discharge is irreversible, the broad peak disappears in the CV curves of the second and third scanning cycles. Generally, LixGe alloy was formed in the voltage range between 0.01 and 0.34 V. The sharp reduction peak at 0.01 V is attributed to lithium intercalation into amorphous carbon. In the corresponding oxidation scanning, the shoulder at 0.25 V is attributed to lithium de-intercalation from amorphous carbon. The broad oxidation peak observed from 0.3 V to 0.7 V can be assigned to the de-alloying of LixGe. The broad peak at 1.1 V can be related to the oxidation from Ge to GeO2. Fig. 3b shows the charge- discharge curves of the first three cycles for the GeO2-C fibers at a constant current density of 50 mA h g-1 with a voltage range of 0.01-3.00 V. The first discharge and charge steps display specific capacities of 1532.56 mA h g-1 and 923.26 mA h g-1 with an initial coulombic efficiency of 60.06%. In the second cycle, coulombic efficiency increase to 96.22% because less SEI is formed and the oxidative phases have been irreversibly reduced during the first discharge, in agreement with the disappearance of the broad peak between 0.5 V and 1.3 V. Fig. 3c shows the cyclic performance of GeO2-C fiber electrode tested at a current density of 50 mA g-1. The GeO2-C fiber electrode exhibits a reversible capacity of 842.08 mA h g-1 at the third cycle and still remains a reversible capacity of 838.93 mA h g-1 even after 100 cycles, which indicates the GeO2-C fiber electrode exhibits excellent cyclic performance. Because the content of GeO2 in composite fibers is 60 wt%, the theoretical capacity attributed to GeO2 is 675.6 mA h g-1 and the extra capacity originates from amorphous carbon produced by carbonization of PAN and PVP in an inert atmosphere, which usually exhibits higher capacity (a reversible capacity of 407.16 mA h g-1 from its cyclic performance in Fig. 3c) than commercial graphite [19]. The amorphous carbon also keeps its reversible capacity very well. But if the GeO2 content further increases to 70 wt%, the cyclic performance of the composite fibers decreases gradually, which indicates the carbon is not enough to buffer the volume change of GeO2 during the discharging and charging processes. Fig. 3d shows the rate performance of GeO2-C fiber electrode. The reversible capacity of the GeO2-C fiber electrode at current density of 200, 400, 600 and 800 mA g-1 is 725.18, 627.41, 534.= and 441.89 mA h g-1, or about 86.=%, 74.88%, 63.80% and 52.74% of the reversible specific capacity at a current density of 50 mA g-1. After the current density return to 50 mA g-1, the composite fiber electrode fully recovers its capacity. The rate performance of amorphous carbon from carbonization of PAN and PVP is similar to that of the composite fibers. Their reversible capacity is 337.61, 285.22, 232.80 and 194.76 mA h g-1, or about 83.32%, 70.39%, 57.45% and 48.06% of the capacity at 50 mA g-1.

|
Download:
|
| Fig. 3.(a) CV curves of GeO2-C fiber anode, (b) galvanostatic charge-discharge voltage profiles of GeO2-C fiber anode, (c) cyclic performance of GeO2-C fiber anode with 30 wt% and 40 wt% carbon, amorphous carbon anode at a current density of 50 mA g-1, (d) rate performance of GeO2-C fiber anode with 40 wt% carbon and amorphous carbon anode at current densities of 50, 200, 400, 600, 800 and 50 mA g-1. | |
{kind=link}
The excellent cyclic performance of the GeO2-C composite fiber electrode can be attributed to the one-dimensional structure with amorphous GeO2 embedding in the carbon matrix [13, 14, 15]. The one-dimensional structure and carbon matrix can release the strain induced by the volume change of GeO2 during the discharging and charging processes and maintain the structural stability of the composite fibers, which may keep the connectivity of the GeO2 particles and improve their conductivity. In addition, the carbon matrix can separate the GeO2 particles, prevent them from aggregating during the electrochemical reaction processes and provide extra capacity for GeO2-C composite fibers [10, 13, 15].
4. ConclusionA transparent, homogeneous and spinnable sol was prepared by adding PANand PVP with a 1:1weight ratio into amixture produced by dropping GeCl4 inDMF and subsequent stirring for several hours. The electrospun fibers with uniform diameters of ~300 nm will transform to amorphous GeO2-C fiberswith the samemorphologies after being calcined at 600 ℃ in Ar atmosphere. The GeO2-C fiber anode exhibits highcapacityandexcellent cyclic performancewith a stable reversible capacity of 838.93 mA h g-1 at a current density of 50 mA g-1 and can retain 446.08mA h g-1 under a current density of 800 mA g-1. The excellent chemical performance is attributed to their one-dimensional structure with GeO2 embedding in the amorphous carbon matrix, which originates from carbonization of PAN and PVP.
AcknowledgmentsThe work was supported by Shanghai Municipal Education Commission (High-energy Beam Intelligent Processing and Green Manufacturing) and Graduate Students Innovation Program of Shanghai University of Engineering Science (No. E1-0903-15- 01040).
| [1] | L. Li, K.H. Seng, C.Q. Feng, H.K. Liu, Z.P. Guo, Synthesis of hollow GeO2 nanostructures, transformation into Ge@C, and lithium storage properties, J. Mater. Chem. A 1(2013) 7666-7672. |
| [2] | K.H. Seng, M.H. Park, Z.P. Guo, H.K. Liu, J. Cho, Catalytic role of Ge in highly reversible GeO2/Ge/C nanocomposite anode material for lithium batteries, Nano Lett. 13(2013) 1230-1236. |
| [3] | Y.M. Lin, K.C. Klavetter, A. Heller, C. Buddie Mullins, Storage of lithium in hydrothermally synthesized GeO2 nanoparticles, J. Phys. Chem. Lett. 4(2013) 999-1004. |
| [4] | J.K. Feng, M.O. Lai, L. Lu, (Ⅰ)nfluence of grain size on lithium storage performance of germanium oxide films, Electrochim. Acta 62(2012) 103-108. |
| [5] | X. Gu, L. Chen, Z.C. Ju, et al., Controlled growth of porous α-Fe2O3 branches on β-MnO2 nanorods for excellent performance in lithium-ion batteries, Adv. Funct. Mater. 23(2013) 4049-4056. |
| [6] | J.Y. Xiang, J.P. Tu, Y.F. Yuan, et al., Electrochemical investigation on nanoflowerlike CuO/Ni composite film as anode for lithium ion batteries, Electrochim. Acta 54(2009) 1160-1165. |
| [7] | T.J. Athauda, J.G. Neff, L. Sutherlin, U. Butt, R.R. Ozer, Systematic study of the structure-property relationships of branched hierarchical TiO2/ZnO nanostructures, ACS Appl. Mater. (Ⅰ)nterfaces 4(2012) 6917-6926. |
| [8] | M.X. Ma, Mesoporous cobalt oxide for largely improved lithium storage properties, Chin. Chem. Lett. 23(2012) 949-952. |
| [9] | S.L. Yang, B.H. Zhou, M. Lei, et al., Sub-100 nm hollow SnO2@C nanoparticles as anode material for lithium ion batteries and significantly enhanced cycle performances, Chin. Chem. Lett. 26(2015) 1293-1297. |
| [10] | Y. Wang, J.Y. Lee, H.C. Zeng, Polycrystalline SnO2 nanotubes prepared via infiltration casting of nanocrystallites and their electrochemical application, Chem. Mater. 17(2005) 3899-3903. |
| [11] | Z.X. Yang, G.D. Du, C.Q. Feng, et al., Synthesis of uniform polycrystalline tin dioxide nanofibers and electrochemical application in lithium-ion batteries, Electrochim. Acta 55(2010) 5485-5491. |
| [12] | L. Qiao, X.H. Wang, L. Qiao, et al., Single electrospun porous NiO-ZnO hybrid nanofibers as anode materials for advanced lithium-ion batteries, Nanoscale 5(2013) 3037-3042. |
| [13] | H.R. Jung, W.J. Lee, Electrochemical characterization of electrospun SnOx-embedded carbon nanofibers anode for lithium ion battery with EXAFS analysis, J. Electroanal. Chem. 662(2011) 334-342. |
| [14] | D. Kim, D. Lee, J. Kim, J. Moon, Electrospun Ni-added SnO2-carbon nanofiber composite anode for high-performance lithium-ion batteries, ACS Appl. Mater. (Ⅰ)nterfaces 4(2012) 5408-5415. |
| [15] | Z.X. Yang, G.D. Du, Z.P. Guo, et al., Easy preparation of SnO2@carbon composite nanofibers with improved lithium ion storage properties, J. Mater. Res. 25(2010) 1516-1524. |
| [16] | hhttp://www.lasurface.com/database/elementxps.phpi |
| [17] | N.R. Murphy, J.T. Grant, L. Sun, et al., Correlation between optical properties and chemical composition of sputter-deposited germanium oxide (GeOx) films, Opt. Mater. 36(2014) 1177-1182. |
| [18] | B. Wang, J.L. Cheng, Y.P. Wu, D. Wang, D.N. He, Electrochemical performance of carbon/Ni composite fibers from electrospinning as anode material for lithium ion batteries, J. Mater. Chem. A 1(2013) 1368-1373. |
| [19] | J. Jin, Z.Q. Shi, C.Y. Wang, The structure and electrochemical properties of carbonized polyacrylonitrile microspheres, Solid State (Ⅰ)onics 261(2014) 5-10. |