 2016, Vol.27
2016, Vol.27 



b. Department of Chemistry, Georgia State University, Atlanta, GA 30303, USA
Protein kinases have become one of the most intensively pursued classes of drug targets with a number of clinical success stories [1]. Receptor protein tyrosine kinases play an important role in signal transduction pathways that regulate cell division and differentiation. Epidermal growth factor receptor tyrosine kinase (EGFR-TK) and the related human epidermal growth factor receptor are among the growth factor receptor kinases that have been identified as important in cancer development [2]. EGFR dependent aberrant signaling is associated with cancer cell proliferation,apoptosis,angiogenesis and metastasis [3]. Anilinoquinazoline- containing compounds,erlotinib (Tarceva®) [3, 4] and gefitinib (Iressa®) [5] have been approved for the treatment of advanced non-small cell lung cancer. They contain the 4-(substituted amino)quinazoline pharmacophoric core that binds to the hinge region of the kinase.
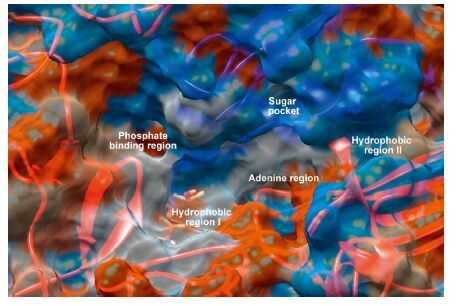
Quinazolines have emerged as a versatile template for inhibitors of a diverse range of receptor tyrosine kinases. The most widely studied of these,is the epidermal growth factor receptor (EGFR) small molecule inhibitor erlotinib,which has been approved for the treatment of non-small cell lung cancer [6, 7]. Subsequent research aimed at further exploration of the SAR of this novel template led to the discovery of highly selective compounds that target EGFR. Benzothiazoles act via competing with ATP for binding at the catalytic domain of EGFR-TK [8]. The ATP-binding site (Fig. 1) has the following features: adenine region,which contains two key hydrogen bonds formed by the interaction of N1 and N6 of the adenine ring. Many potent inhibitors use one of these hydrogen bonds; sugar pocket,which is a hydrophilic region; hydrophobic regions and channels,which play an important role in inhibitor selectivity and binding affinity; and phosphate binding region,which is largely solvent exposed and can be used for improving inhibitor selectivity [9].

|
Download:
|
| Fig. 1.The molecular surface representation of the ATP-binding site that consists of the adenine region,hydrophobic regions I and II,sugar pocket and phosphate binding region [9]. | |
Erlotinib binds to the active conformation of EGFR and exploits the threonine gatekeeper (Thr-790) to penetrate in the EGFR back cleft through this gate. The crystal structure of EGFR in complex with erlotinib (PDB code: 1M17) (http://www.rcsb.org/pdb/home/home.do) showed the binding mode of erlotinib (Fig. 2). N1 of the quinazoline core accepts a hydrogen bond from the amide nitrogen of Met-769 (numbering used in PDB code: 1M17) in the hinge region. Moreover,the acetylene moiety at the 3-position of the phenyl ring is directed into the hydrophobic region I,significantly improving the compound selectivity,whereas substituents at 6- and 7-positions of the quinazoline ring extend into the entrance regions.

|
Download:
|
| Fig. 2.The 3D and 2D representation of EGFR crystal structure in complex with erlotinib (PDB code: 1M17). (A) The 3D representation of EGFR crystal structure in complex with erlotinib; (B) The 2D representation of EGFR crystal structure in complex with erlotinib. | |
Benzothiazole derivatives constitute an important class of therapeutic agents in medicinal chemistry. Literature survey revealed that this nucleus is associated with diverse pharmacological effects,including antitumor [10, 11, 12, 13, 14, 15, 16, 17] and antioxidant [18, 19] activities. In addition,benzothiazole Schiff bases were reported to have antitumor activity [20]. Taking all these findings into consideration,we present herein a new subfamily of compounds containing the benzothiazole core for evaluation of their antitumor activity. Our strategy is directed toward designing a variety of ligands,which are structurally similar to the basic skeleton,4-anilinoquinazoline of tinibs (erlotinib and gefitinib) with diverse chemical properties (Fig. 3). Accordingly,we replaced quinazoline ring with benzothiazole since both rings are isosteric with the adenine portion of ATP and can mimic the ATPcompetitive binding regions of EGFR-TK.

|
Download:
|
| Fig. 3.Reported antitumor quinazolines and proposed compounds. | |
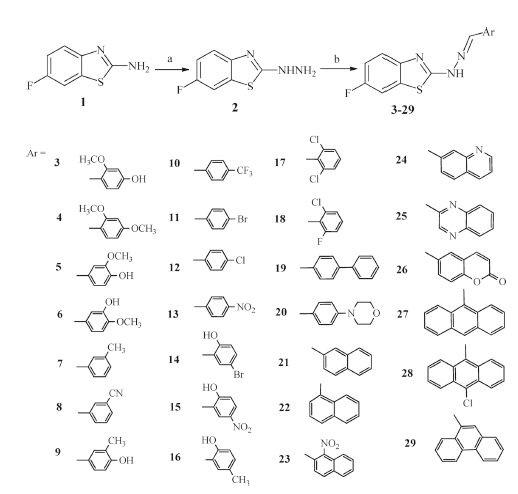
A general approach for the synthesis of the designed compounds is outlined in Scheme 1. The starting compound,2- amino-6-fluorobenzothiazole (1) was reacted with hydrazine hydrate in refluxing ethylene glycol in the presence of hydrochloric acid to produce the hydrazine derivative 2 [21]. Reaction of compound 2 with the appropriate aromatic aldehyde in ethanol under microwave irradiation gave the corresponding Schiff bases 3-29. The synthesis information and characterization data of target compounds are deposited in Supporting information. The antitumor screening of compounds 3-29 against cervical cancer (Hela) and kidney fibroblast cancer (COS-7) cell lines was carried out adopting the MTT assay [22, 23, 24] and using doxorubicin as a reference antitumor agent. In addition,structure based pharmacophore mapping,molecular docking,protein-ligand interaction,fingerprints and binding energy calculations were employed in a virtual screening strategy to identify the interaction between the compounds and the active site of the putative target,EGFR-TK.

|
Download:
|
| Scheme 1.Reagents and reaction conditions: (a) hydrazine hydrate,hydrochloric acid,ethylene glycol,reflux,5 h; (b) aromatic aldehydes,ethanol,microwave (20 W),80 ℃,10 min. | |
The structures of all the synthesized compounds were confirmed by IR,1H NMR,13C NMR and HRMS. 1H NMR spectra of compounds 3-29 showed two characteristic singlets at δ 7.98- 9.35 and 11.40-13.00 for CH=N and NH,respectively. The antitumor activity of the synthesized compounds 3-29 against cervical cancer (Hela) and kidney fibroblast cancer (COS-7) cell lines was evaluated. Results are expressed in IC50 (mmol/L) and presented in Table 1. As shown in Table 1,compounds 3,14,19,27 and 28 have promising activity against Hela cell line with IC50 values of 2.41,3.06,6.46,2.22 and 6.25 mmol/L,respectively,in comparison to doxorubicin as a reference antitumor agent with IC50 value of 2.05 mmol/L. In addition,compound 3 displayed excellent activity against COS-7 cell line with IC50 value of 4.31 mmol/L in comparison to doxorubicin with IC50 value of 3.04 mmol/L.
|
|
Table 1 In vitro antitumor activity of compounds 3-29 against cervical cancer (Hela) and kidney fibroblast cancer (COS-7) cell lines. |

Structure activity correlation of compounds 3-29 based on the tested cell lines,cervical cancer (Hela) and kidney fibroblast cancer (COS-7) cell lines,is discussed. The presence of 2-(4-hydroxy-2- methoxybenzylidene)hydrazino moiety at the 2-position of benzothiazole nucleus greatly enhanced the activity against cervical cancer (Hela) and kidney fibroblast cancer (COS-7) cell lines (compound 3). On the other hand,replacement of 4-hydroxy substituent in compound 3 with 4-methoxy resulted in decreased activity against both cell lines (compound 4). Furthermore,changing the positions of the hydroxy and methoxy substituents on the benzylidene moiety led to decreased activity against both cell lines (compounds 5 and 6). In addition,the presence of 2-(3- methylbenzylidene)hydrazino moiety improved the hydrophobic interaction with the receptor and resulted in considerable activity against both cell lines (compound 7). On the other hand,the presence of 2-(4-hydroxy-3-methylbenzylidene)hydrazino moiety showed decreased activity against both cell lines (compound 9).
Introduction of trifluoromethyl substituent at the para position of the benzylidene moiety increased the activity against Hela cell line compared to the other electron-withdrawing substituents (Br,Cl,NO2) (compound 10 versus 11,12 and 13),whereas the presence of chloro substituent at the para position of the benzylidene moiety increased the activity against COS-7 cell line compared to the other electron-withdrawing substituents (CF3,Br,NO2) (compound 12 versus 10,11 and 13). Moreover,the presence of 2-(5-bromo-2- hydroxybenzylidene)hydrazino moiety enhanced the activity against Hela cell line (compound 14). Replacement of 5-bromo substituent with 5-nitro or 5-methyl substituents resulted in decreased activity against the same cell line (compounds 15 and 16). In addition,the presence of 2-(2,6-dichlorobenzylidene)hydrazino moiety contributed to the considerable activity of compound 17 against Hela cell line. On the other hand,replacement of the 2-(2,6-dichlorobenzylidene)hydrazino moiety with the 2-(2-chloro-6-fluorobenzylidene)hydrazino counterpart resulted in decreased activity against the same cell line (compound 18).
The presence of 2-(naphthalen-2-ylmethylene)hydrazino moiety at the 2-position of benzothiazole scaffold improved the activity against Hela cell line compared to the 2-(naphthalen-1- ylmethylene)hydrazino moiety (compound 21 versus 22). On the other hand,the presence of nitro substituent at 1-position of naphthalene ring of 2-(naphthalen-2-ylmethylene)hydrazino moiety led to decreased activity against the same cell line (compound 23 versus 21). Replacement of the 2-(naphthalen-2-ylmethylene)- hydrazino moiety with the 2-(1,10-biphenyl-4-ylmethylene)hydrazino counterpart improved the activity against both cell lines (compound 19). On the other hand,replacement of the 1,10- biphenyl moiety with 4-(morpholin-4-yl)phenyl decreased the activity against both cell lines (compound 20). Furthermore,replacement of the 2-(naphthalen-2-ylmethylene)hydrazino moiety with the 2-(quinolin-7-ylmethylene)hydrazino or 2-(quinoxalin- 2-ylmethylene)hydrazino counterparts decreased the activity against Hela cell line (compound 21 versus 24 and 25). On the other hand,introduction of 2-(2-oxo-2H-chromen-6-yl)hydrazino counterpart improved the activity against the same cell line (compound 26). Attempts to improve hydrophobic interaction with the target receptor by adding an extra phenyl ring via replacing the 2- (naphthalen-2-ylmethylene)hydrazino moiety with the 2-(anthracen- 9-ylmethylene)hydrazino counterpart greatly enhanced the activity against Hela cell line (compound 27). Trials to introduce an extra site for hydrophobic interaction in compound 27 by the addition of chloro substituent at the 10-position of the anthracene ring decreased the activity against the same cell line (compound 28). In addition,the presence of 2-(phenanthren-9-ylmethylene)- hydrazino moiety decreased the activity against Hela cell line but improved the activity against COS-7 cell line (compound 29).
3.2. Molecular modeling and computational studiesOveractivation of receptor tyrosine kinase (RTK) signaling pathways is strongly associated with carcinogenesis. Thus,it is becoming increasingly clear that impaired deactivation of RTKs may be an oncogenic driver of cancer [25]. On this basis,Computer- Aided Drug Design (CADD) tools were used to identify the interaction between the newly synthesized compounds and the active site of EGFR-TK in comparison to erlotinib as a reference EGFR-TK inhibitor. Kinase inhibitors should contain the following features to gain selectivity and potency [26]: A portion that closely mimics the ATP molecule and one to three hydrogen bonds with the amino acids located in the hinge region of the target kinases,as in erlotinib [27],lapatinib [28] and gefitinib [29]. An additional hydrophobic binding site (allosteric site),which is directly adjacent to the ATP-binding site,as in imatinib [30] and sorafenib [31]. However,other mechanism could be achieved through binding outside the ATP-binding site at an allosteric site [32] and by forming an irreversible covalent bond to the kinase active site [33, 34]. In the present work,erlotinib binding mode to EGFR-TK was studied and the design of the newly synthesized compounds is based on the essential chemical features required for erlotinib binding affinity to EGFR-TK.
3.2.1. Similarity-based virtual screeningSimilarity methods may be the simplest and most widely used tools for ligand-based virtual screening of chemical databases,where functionally similar molecules are sought by searching molecular databases for structurally similar molecules. These methods can be categorized as 2D and 3D similarity methods. However,the most common approaches are based on the 2D fingerprints,with the similarity between a reference structure and a database structure.
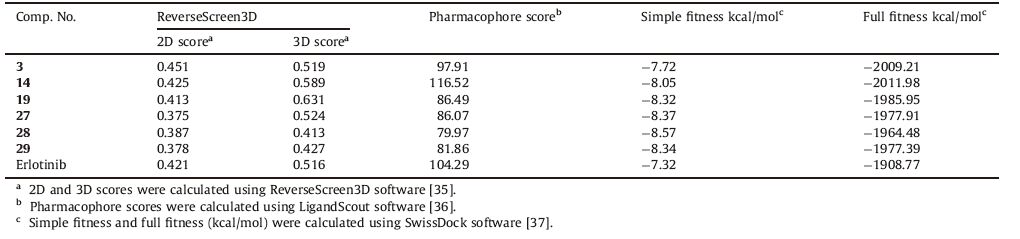
The most active compounds,3,14,19,27,28 and 29 in Standard Delay Format (SDF) were submitted to the ReverseScreen3D server [35],the server uses a reverse virtual screening (VS) method called ReverseScreen3D. It is a 2D fingerprint-based method to select a ligand template from each unique binding site of each protein with a target database. The target database contains only the structurally determined bioactive conformations of known ligands. The 2D comparison is followed by a 3D structural comparison to the selected query ligand using a geometric matching method in order to priotrize each target binding site in the database. The output in the form of a list of the 2D and 3D scores for protein tyrosine kinase (cluster no. 14836) is listed in Table 2. Compounds 3,14,19 and 27 with promising antitumor activity,displayed the highest 3D score values of 0.519,0.589,0.631 and 0.524,respectively.
|
|
Table 2 Results of molecular docking analysis of the most active compounds in the EGFR-TK active site. |
3D Pharmacophore designing methods take into account both the 3-dimensional structures and binding modes of receptors and inhibitors in order to identify regions that are favorable for specific receptor-inhibitor interaction. The description of the receptor- inhibitor interaction pattern is determined by a correlation between the characteristic properties of the inhibitors and their biochemically determined enzymatic activity.
LigandScout,a program that allows the automatic construction and visualization of 3D pharmacophore from structural data of protein-ligand complexes,was used in this study to create a pharmacophore for the mode of action of erlotinib,which prevents the activation of EGFR kinase [36]. The model (Fig. 4) was created by automatically overlaying pharmacophoric features gathered from the crystal structure of EGFR kinase domain (PDB ID: 1M17) in complex with erlotinib (http://www.rcsb.org/pdb/home/home.do).

|
Download:
|
| Fig. 4.(A) The crystal structure of EGFR kinase domain (PDB ID: 1M17) in complex with erlotinib was obtained from the protein data bank (PDB); (B) LigandScout 3D proposed docking pose of erlotinib in the ATP binding site of EGFR kinase domain; (C) The 3D pharmacophore of erlotinib (in ball and stick representation); The pharmacophore color coding is red for hydrogen bond acceptors green for hydrogen bond donors and yellow for hydrophobic regions; (D) The final 3D pharmacophore model for EGFR kinase domain. | |
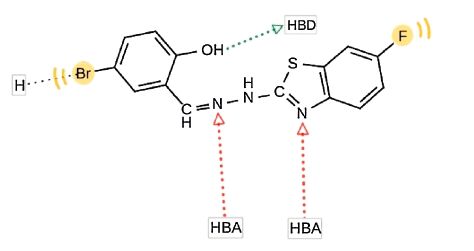
The investigated pharmacophoric features included hydrogen bond donors and acceptors as directed vectors,positive and negative ionizable regions as well as lipophilic areas that are represented by spheres. According to the pharmacophore generated by LigandScout,the minimal structural requirements for antitumor activity consist of a hydrophobic region attached to a heterocyclic ring that fits into the ATP-binding site,two hydrogen bond acceptors and one hydrogen bond donor. The 3D alignment of the pharmacophoric features of each of the synthesized compounds and the 3D pharmacophore of erlotinib binding pose showed that these compounds possess similar pharmacophoric features required for activity. The pharmacophore score listed in Table 2 was calculated for the alignment of compounds 3,14,19,27,28 and 29 into the 3D pharmacophore of erlotinib binding pose generated by LigandScout. This score reflects the similarity of the compounds to the reference pharmacophore. Compound 14 showed the highest pharmacophore score value of 116.52. The detailed 2D mapping of the pharmacophore model with the structural features of compound 14 is depicted in Fig. 5.

|
Download:
|
| Fig. 5.The 2D representation of the structural features of compound 14 that can be aligned with the pharmacophore hypothesis. HBA,hydrogen bond acceptor; HBD,hydrogen bond donor; H,hydrophobic center. | |
The crystal structure of EGFR kinase domain (PDB ID: 1M17) in complex with an irreversible inhibitor was obtained from the protein data bank (PDB) (http://www.rcsb.org/pdb/home/home.do). Docking simulation was done by using the SwissDock software [37]. All the conformers were virtually docked at the defined cavity of the receptor. The docking scores of the best conformers for each ligand are listed (Table 2). The ligand forming the most stable drug receptor complex is the one having the lowest docking score value. The six active compounds are evaluated using two scoring functions: simple fitness and full fitness. Simple fitness is a fast and efficient method to evaluate the individual binding modes but neglects the solvent effect and is used to drive the search. Simultaneously,clusters of binding modes are evaluated by the more selective yet slower,full fitness method,which accounts for the solvation free energy. Compound 14 showed a relatively low full fitness score value of -2011.98 kcal/mol. 3D Interactions of compound 14 with the binding site of EGFR-TK are shown in Fig. 6.

|
Download:
|
| Fig. 6.3D Interactions of compound 14 with EGFR-TK binding site. The atoms are colored as following: red for oxygen atoms,blue for nitrogen atoms,yellow for sulfur atoms,white for hydrogen atoms,cyan for carbon atoms and green for chlorine atoms. | |
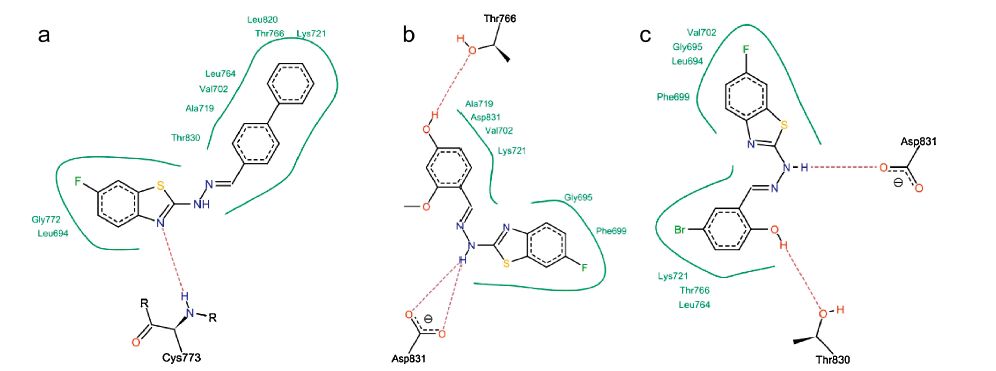
Analysis of the docking results (Table 2) using the Lead IT software revealed that the main interaction forces of the candidate compounds with the EGFR-TK active site are hydrophobic in nature [38]. Enhancement of the antitumor activity of compound 19 may be explained by the improved hydrophobic interactions with EGFR-TK active site as illustrated in Fig. 7a. The important residues in the hydrophobic regions that interact with the hit compounds are (Phe-699,Ala-719,Val-702,Lys-721 and Asp-831). The docking results showed that hydrogen bonding interactions of the newly synthesized compounds with EGFR-TK binding site greatly enhanced the affinity toward the enzyme. Analysis of binding mode revealed that compounds 3 and 14 are involved in the formation of two hydrogen bonds with the EGFR-TK active site,which indicates a correlation between the hydrogen bonding interactions and the antitumor activity (Fig. 7b and c).

|
Download:
|
| Fig. 7.2D Interactions of compounds 19 (a),3 (b) and 14 (c) with EGFR-TK binding site. Dashed lines represent hydrogen bonds. Hydrophobic interactions are shown by green solid lines. | |
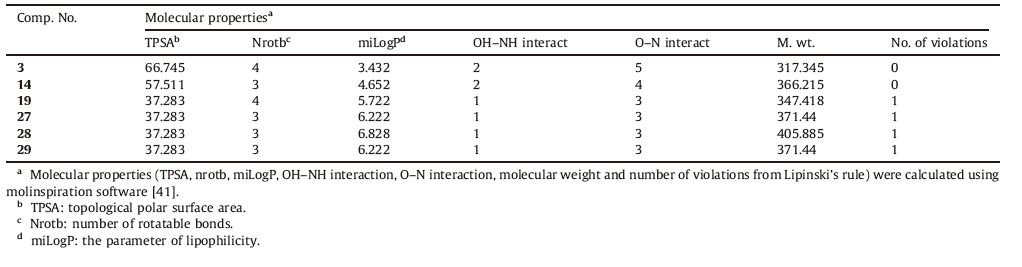
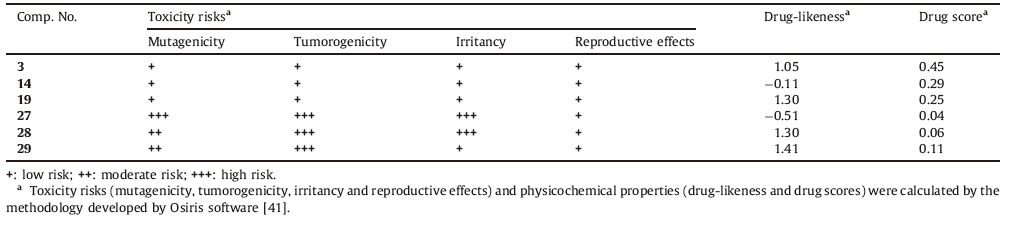
Drug-likeness is a complex balance of various structural features that determine whether a particular molecule is similar to the known drugs or not. It generally means "molecules that contain functional groups and/or have physical properties consistent with most of the known drugs". Hydrophobicity,molecular size,flexibility and presence of various pharmacophoric features are the main physical properties that influence the behavior of molecules in a living organism. Computational chemists have a wide array of tools and approaches available for the assessment of molecular diversity. Diversity analysis has been shown to be an important ingredient in designing drugs. So,computational sensitivity analysis and structural analysis have been used to study the drug-likeness of the candidate drugs. As good bioavailability can be achieved with an appropriate balance between solubility and partitioning properties,the six active compounds,3,14,19,27,28 and 29 were analyzed for the prediction of Lipinski’s rule of five [39] as well as other properties (Tables 3 and 4).
3.3.1. Molinspiration calculationsAs a part of our study; the compliance of compounds to the Lipinski’s rule of five was evaluated [39]. This simple rule is based on the observation that most drugs that passed phase 2 clinical trials have molecular weight of 500 or less,clogP values lower than 5,hydrogen bond donors fewer than 5 and hydrogen bond acceptors fewer than 10. In addition,topological polar surface area (TPSA) and number of rotatable bonds have also been linked to drug bioavailability [40].
Molecular properties (TPSA,nrotb,miLogP,OH-NH interaction,O-N interaction,molecular weight and number of violations from Lipinski’s rule) of the six active compounds,3,14,19,27,28 and 29 were calculated using the molinspiration software [41] (Table 3). Topological polar surface area (TPSA) and lipophilicity (logP) values are two important properties for the prediction of oral bioavailability of drug molecules [41, 42, 43, 44]. TPSA is calculated based on the methodology published by Ertl et al. [44] as the surface areas that are occupied by oxygen and nitrogen atoms and by hydrogen atoms attached to them. Thus,it is closely related to the hydrogen bonding potential of a compound [41, 42, 43, 44]. TPSA has been shown to be a very good descriptor characterizing drug absorption,including intestinal absorption,bioavailability and blood-brain barrier penetration. Molecules with TPSA values of 140Å 2 or more are expected to exhibit poor intestinal absorption [40]. Results shown in Table 3 indicated that all the analyzed compounds have TPSA values < 140Å 2; thus,they are expected to have good intestinal absorption. Molecules with more than 10 rotatable bonds may have problems with bioavailability [40]. All the analyzed compounds have 3 or 4 rotatable bonds and they might not have problems with bioavailability (Table 3). MiLogP is calculated using the methodology developed by Molinspiration as a sum of fragment-based contributions and correction factors (http://www.molinspiration.com) (Table 3). It has been shown that for a compound to have a reasonable probability of being well absorbed,miLogP value must be in the range of -0.4 to +5.6 [40]. On this basis,compounds 3 and 14 were found to have miLogP values within the acceptable criteria. It is worth mentioning that all the analyzed compounds have one or zero violation of Lipinski’s rule and they are expected to have reasonable oral absorption.
|
|
Table 3 Topological polar surface area,number of rotatable bonds and calculated Lipinski’s rule of five for the most active compounds. |
Toxicity risks (mutagenicity,tumorogenicity,irritancy and reproductive effects) and physicochemical properties (druglikeness and drug score) of the synthesized compounds were calculated by the methodology developed by Osiris [41]. The toxicity risk predictor locates fragments within a molecule that indicate a potential toxicity risk. Toxicity risk alerts are an indication that the drawn structure may be harmful concerning the risk category specified. From the data presented in Table 4,it is obvious that compounds 3,14 and 19 are expected to be nonmutagenic and non-tumorigenic. Compounds 3,14,19 and 29 are expected to be non-irritant. In addition,all analyzed compounds were found to have non-reproductive effects.
|
|
Table 4 Toxicity risks,drug-likeness and drug score for the most active compounds. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Drug-likeness is defined as a complex balance of various molecular properties and structural features that indicates whether a particular molecule is similar to the known drugs or not [45]. Osiris program was used in calculating the fragmentbased drug-likeness of the synthesized compounds,where a positive value indicates that the designed molecule contains fragments that are frequently present in commercial drugs. Results shown in Table 4 indicated that compounds 3,19,28 and 29 have positive drug-likeness values. The drug score combines druglikeness,miLogP,solubility,molecular weight and toxicity risks in one handy value that may be used to judge the compound’s overall potential to qualify for a drug [41]. A value of 0.5 or more makes the compound a promising lead for future development of safe and efficient drugs. The overall drug score values for the synthesized compounds were calculated (Table 4).
4. ConclusionCompounds 3,14,19,27 and 28 are the most active antitumor agents in this study against cervical cancer (Hela) cell line. In addition,compounds 3 and 29 are the most active members against kidney fibroblast cancer (COS-7) cell line. From these results we can conclude that the actual antitumor activity of compounds 3,19,28 and 29 is correlated to the drug-likeness prediction results as these compounds have positive drug-likeness values. These encouraging preliminary results of biological screening of the newly synthesized compounds could offer a good framework toward the discovery of new potent antitumor agents.
AcknowledgmentsThe authors thank Professor Binghe Wang at Georgia State University,USA,for providing all required facilities and carrying out the spectral and elemental analyses as well as MTT assay for antitumor screening.
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.12033.
| [1] | Q. Liu, Y. Sabnis, Z. Zhao, et al., Developing irreversible inhibitors of the protein kinase cysteinome, Chem. Biol. 21(2013) 146-159. |
| [2] | L.V. Peng-Cheng, C.F. Zhou, J. Chen, et al., Design, synthesis and biological evaluation of thiazolidinone derivatives as potential EGFR and HER-2 inhibitors, Bioorg. Med. Chem. Lett. 18(2010) 314-319. |
| [3] | M. Reck, N.V. Zandwijk, C. Gridelli, et al., Erlotinib in advanced non-small cell lung cancer:efficacy and safety findings of the global phase (Ⅰ)V Tarceva lung cancer survival treatment study, J. Thorac. Oncol. 5(2010) 1616-1622. |
| [4] | J. Smith, Erlotinib:small-molecule targeted therapy in the treatment of non smallcell lung cancer, Clin. Ther. 27(2005) 1513-1534. |
| [5] | K. Tamura, M. Fukuoka, Gefitinib in non-small cell lung cancer, Expert Opin. Pharmacother. 6(2005) 985-993. |
| [6] | P. Ballard, R.H. Bradbury, C.S. Harris, et al., (Ⅰ)nhibitors of epidermal growth factor receptor tyrosine kinase:optimisation of potency and in vivo pharmacokinetics, Bioorg. Med. Chem. Lett. 16(2006) 4908-4912. |
| [7] | M. Ranson, Epidermal growth factor receptor tyrosine kinase inhibitors, Br. J. Cancer 90(2004) 2250-2255. |
| [8] | M.N. Noolvi, H.M. Patel, M. Kaur, Benzothiazoles:search for anticancer agents, Eur. J. Med. Chem. 54(2012) 447-462. |
| [9] | D. Fabbro, S. Ruetz, E. Buchdunger, et al., Protein kinases as targets for anticancer agents:from inhibitors to useful drugs, Pharmacol. Ther. 93(2002) 79-98. |
| [10] | X.H. Shi, Z. Wang, Y. Xia, et al., Synthesis and biological evaluation of novel benzothiazole-2-thiol derivatives as potential anticancer agents, Molecules 17(2012) 3933-3944. |
| [11] | H.S. Elzahabi, Synthesis, characterization of some benzazoles bearing pyridine moiety:search for novel anticancer agents, Eur. J. Med. Chem. 46(2011) 4025-4034. |
| [12] | S. Saeed, N. Rashid, P.G. Jones, M. Ali, R. Hussain, Synthesis, characterization and biological evaluation of some thiourea derivatives bearing benzothiazole moiety as potential antimicrobial and anticancer agents, Eur. J. Med. Chem. 45(2010) 1323-1331. |
| [13] | W.P. Hu, Y.K. Chen, C.C. Liao, et al., Synthesis, and biological evaluation of 2-(4-aminophenyl)benzothiazole derivatives as photosensitizing agents, Bioorg. Med. Chem. 18(2010) 6197-6207. |
| [14] | Y.A. Al-Soud, H.H. Al-Sa'doni, B. Saeed, et al., Synthesis and in vitro antiproliferative activity of new benzothiazole derivatives, ARK(Ⅰ)VOC xv (2008) 225-238. |
| [15] | G.M. Catriona, W. Geoffrey, C.P. Jean, et al., Antitumor benzothiazoles. 26. 2-(3,4-dimethoxyphenyl)-5-fluorobenzothiazole (GW 610 NSC 721648), a simple fluorinated 2-arylbenzothiazole, shows potent and selective inhibitory activity against lung, colon and breast cancer cell lines, J. Med. Chem. 49(2006) 179-185. |
| [16] | E. Brantley, S. Antony, G. Kohlhagen, et al., Anti-tumor drug candidate 2-(4-amino-3-methylphenyl)-5-fluorobenzothiazole induces single-strand breaks and DNA-protein cross-links in sensitive MCF-7 breast cancer cells, Cancer Chemother. Pharmacol. 58(2006) 62-72. |
| [17] | C.J. Lion, C.S. Matthews, G. Wells, et al., Antitumour properties of fluorinated benzothiazole-substituted hydroxycyclohexa-2,5-dienones (‘quinols’), Bioorg. Med. Chem. Lett. 16(2006) 5005-5008. |
| [18] | N. Karali, O. Güzel, N. Ozsoy, S. Ozbey, A. Salman, Synthesis of new spiroindolinones incorporating a benzothiazole moiety as antioxidant agents, Eur. J. Med. Chem. 45(2010) 1068-1077. |
| [19] | D. Cressier, C. Prouillac, P. Hernandez, et al., Synthesis, antioxidant properties and radioprotective effects of new benzothiazoles and thiadiazoles, Bioorg. Med. Chem. 17(2009) 5275-5284. |
| [20] | S.E. Etaiw, D.M. Abd El-Aziz, E.H. Abd El-Zaher, E.A. Ali, Synthesis, spectral, antimicrobial and antitumor assessment of Schiff base derived from 2-aminobenzothiazole and its transition metal complexes, Spectrochim. Acta A:Mol. Biomol. Spectrosc. 79(2011) 1331-1337. |
| [21] | P. Yadav, D. Chauhan, N.K. Sharma, S. Singhal, 2-Substituted hydrazino-6-fluoro-1,3-benzothiazole:synthesis and characterization of new novel antimicrobial agents, (Ⅰ)nt. J. ChemTech Res. 2(2010) 1209-1213. |
| [22] | T. Mosmann, Rapid colorimetric assay for cellular growth and survival:application to proliferation and cytotoxicity assays, J. (Ⅰ)mmunol. Methods 65(1983) 55-63. |
| [23] | F. Denizot, R. Lang, Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability, J. (Ⅰ)mmunol. Methods 89(1986) 271-277. |
| [24] | D. Gerlier, T. Thomasset, Use of MTT colorimetric assay to measure cell activation, J. (Ⅰ)mmunol. Methods 94(1986) 57-63. |
| [25] | G.B. Kristi, S. Thomas, S. Herald, Defective down regulation of receptor tyrosine kinases in cancer, Eur. Mol. Biol. Organ. 23(2004) 2707-2712. |
| [26] | J. Zhang, P.L. Yang, N.S. Gray, Targeting cancer with small molecule kinase inhibitors, Nat. Rev. Cancer 9(2009) 28-39. |
| [27] | B. Liu, B. Bernard, J.H. Wu, (Ⅰ)mpact of EGFR point mutations on the sensitivity to gefitinib:insights from comparative structural analyses and molecular dynamics simulations, Proteins 65(2006) 331-346. |
| [28] | Z.A. Wainberg, A. Anghel, A.J. Desai, et al., Lapatinib, a dual EGFR and HER2 kinase inhibitor, selectively inhibits HER2-amplified human gastric cancer cells and is synergistic with trastuzumab in vitro and in vivo, Clin. Cancer Res. 16(2010) 1509-1519. |
| [29] | C. Yun, T.J. Boggon, Y. Li, et al., Structures of lung cancer-derived EGFR mutants and inhibitor complexes:mechanism of activation and insights into differential inhibitor sensitivity, Cancer Cell 11(2007) 217-227. |
| [30] | M. Cherry, D.H. Williams, Recent kinase inhibitor X-ray structures:mechanisms of inhibition and selectivity insights, Curr. Med. Chem. 11(2004) 663-673. |
| [31] | S. Whittaker, R. Kirk, R. Hayward, et al., Gatekeeper mutations mediate resistance to BRAF-targeted therapies, Sci. Transl. Med. 2(2010) 35-41. |
| [32] | E. Weisberg, P.W. Manley, S.W. Cowan-Jacob, A. Hochhaus, J.D. Griffin, Second generation inhibitors of BCR-ABL for the treatment of imatinib resistant chronic myeloid leukemia, Nat. Rev. Cancer 7(2007) 345-356. |
| [33] | S. Sridhar, L. Seymour, F.A. Shepherd, (Ⅰ)nhibitors of epidermal growth factor receptors:a review of clinical research with a focus on non small-cell lung cancer, Lancet Oncol. 4(2003) 397-406. |
| [34] | F.A. Sharma, R. Sharma, T. Tyagi, Receptor tyrosine kinase inhibitors as potent weapons in war against cancers, Curr. Pharm. Des. 15(2009) 758-776. |
| [35] | S.L. Kinnings, R.M. Jackson, ReverseScreen3D:a structure-based ligand matching method to identify protein targets, J. Chem. (Ⅰ)nf. Model. 51(2011) 624-634. |
| [36] | G. Wolber, T. Langer, LigandScout:3D pharmacophores derived from proteinbound ligands and their use as virtual screening filters, J. Chem. (Ⅰ)nf. Comput. Sci. 45(2005) 160-169. |
| [37] | A. Grosdidier, V. Zoete, O. Michielin, SwissDock, a protein-small molecule docking web service based on EADock DSS, Nucleic Acids Res. 39(2011) W270-W277. |
| [38] | P. Maass, T. Schulz-Gasch, M. Stahl, M. Rarey, ReCore:a fast and versatile method for scaffold hopping based on small molecule crystal structure conformations, J. Chem. (Ⅰ)nf. Model. 47(2007) 390-399. |
| [39] | C.A. Lipinski, F. Lombardo, B.W. Dominy, P.J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev. 46(2001) 3-26. |
| [40] | D.F. Veber, S.R. Johnson, H.Y. Cheng, et al., Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem. 45(2002) 2615-2623. |
| [41] | A. Jarrahpour, J. Fathi, M. Mimouni, et al., Petra, Osiris and molinspiration (POM) together as a successful support in drug design:antibacterial activity and biopharmaceutical characterization of some azo Schiff bases, Med. Chem. Res. 19(2011) 1-7. |
| [42] | A. Parvez, M. Jyotsna, M.H. Youssoufi, T. Ben Hadda, Theoretical calculations and experimental verification of the antibacterial potential of some monocyclic betalactames containing two synergetic buried antibacterial pharmacophore sites, Phosphorus Sulfur Silicon Relat. Elem. 7(2010) 1500-1510. |
| [43] | A. Parvez, J. Meshram, V. Tiwari, et al., Pharmacophores modeling in terms of prediction of theoretical physicochemical properties and verification by experimental correlations of novel coumarin derivatives produced via Betti's protocol, Eur. J. Med. Chem. 45(2010) 4370-4378. |
| [44] | P. Ertl, B. Rohde, P. Selzer, Fast calculation of molecular polar surface area (PSA) as a sum of fragment-based contributions and its application to the prediction of drug transport properties, J. Med. Chem. 43(2000) 3714-3717. |
| [45] | O. Ursu, A. Rayan, A. Goldblum, T. Oprea, Understanding drug-likeness, W(Ⅰ)REs Comput. Mol. Sci. 1(2011) 760-781. |