 2016, Vol.27
2016, Vol.27 



Since du Vigneaud published the first solution-phase synthesis of oxytocin in 1953 [1],it has been found that peptides participate in various biocatalytic processes [2]. With assistance of recent advances in synthetic [3] and drug delivery technologies [4],peptide-based anti-cancer [5],anti-diabetic [6],anti-microbial [7],anti-fungal [8] drugs and intrinsic hormonal analogues have been discovered [9]. Therefore,it is imperative to develop large scale synthetic approaches for the production of complex peptides.
Traditional solution-phase methodologies are the most widely used approaches for industrialproduction of approved peptide-based pharmaceuticals. In general,they have no scale limitations,and each step can be monitored precisely. However,significant shortcomings of solution-phase methodologies,such as consumption of a large amount of organic solvents,tedious protection/de-protection steps and difficult purification processes,still hamper the development of scale-up routes of peptide syntheses. Solid-phase methodologies,in which the peptides of relatively complex sequences could be synthesized using automated and rapid synthesis/workup procedures,were pioneered by BruceMerrifield to overcome the problems in traditional solution-phase methodologies [10]. However,after decades of optimization,shortcomings of solid-phase synthesis still limited its applications in commercial-scale manufacture. For example,lack of scalability,inadequate in-process controls and low purity of the final products still need improvement [11]. Alternative approaches consisting of soluble polymer-tagged liquid-phase reactions have also been developed to ensure both high reaction rate and real time reaction monitoring [12]. In this method,peptide syntheses could be performed in the solution phase but work-up is accomplished in a manner similar to solid-phase synthesis where most of the excess organic reagents and activated amino acids are removed by simple solidification and filtration procedures.However,the high-cost of soluble polymers,the time-consuming process of solidification/crystallization in each coupling cycle and the inherent scale limitation as observed in solid-phase methodologies represent serious challenges for large scale synthesis of peptides.
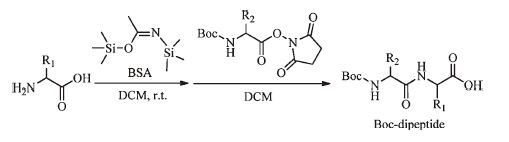
Here we describe a simplified dipeptide synthesis strategy using the N-hydroxysuccinimide (NHS) ester of amino acids and a silylation agent N,O-bis(trimethylsilyl)acetamide (BSA) as a coupling agent (Fig. 1) The coupling reactions between NHS esters and amines occur efficiently under mild conditions with BSA only and no additional acid/base is required. Besides,excessive BSA and other byproducts are water soluble or hydrolysable [13] and easy to remove by simple water-washing at the purification stage. Consequently,less racemization,fewer units of operation,and simpler purification processes can be achieved compared to other solution-phase methodologies.

|
Download:
|
| Fig. 1.Synthesis of t-butyloxycarbonyl (Boc)-protected dipeptide using BSA and NHS ester as coupling agents. | |
All reactions were performed under a nitrogen atmosphere using anhydrous techniques unless otherwise noted. 1H NMR (300 MHz) on a Varian Mercury 300 spectrometer was recorded in DMSO-d6 or CDCl3. Chemical shifts are reported in δ (ppm) units relative to the internal standard tetramethylsilane (TMS). All the reactions were monitored by thin-layer chromatography (TLC) analysis on pre-coated silica gel G plates at 254 nm under UV lamp or HPLC analysis.
2.1. General procedure for the preparation of N-Boc protected dipeptideUnder argon protection,BSA (2.2 equiv.) was added to a solution of amino acid (1.1 equiv.) in anhydrous dichloromethane. After the mixture was stirred for 1-8 h at 23 ℃,a solution of N-Boc protected NHS ester (1 equiv.) in dichloromethane was added. The reaction mixture was stirred at 23 ℃ under argon until all active ester was consumed as judged by TLC analysis. The reaction mixture was washed with brine,dried over Na2SO4 and concentrated in vacuo to provide a white solid. The isolated product was recrystallized from diethyl ether/n-hexane to yield the targeted dipeptide as a white solid.
N-Boc-L-phenylalanine-L-proline (Boc-Phe-Pro-OH): ESI-MS (m/z): 363.2 [M + H]+; 307.1 (M-(CH3)2C = CH2). 1H NMR (400 MHz,CDCl3): δ 7.26 (m,5H),5.39 (d,1H,J = 8.6 Hz),4.69- 4.49 (m,2H),3.63-3.52 (m,1H),3.01 (m,3H),2.28-2.16 (m,1H),2.10-1.98 (m,1H),1.86 (m,2H),1.39 (s,9H).
N-Boc-L-alanine-L-proline (Boc-Ala-Pro-OH): ESI-MS (m/z): 287.2 [M + H]+; 231.1 (M-(CH3)2C = CH2). 1H NMR (400 MHz,CDCl3): δ 4.60 (dd,1H,J = 8.1,3.9 Hz),4.49 (s,1H),3.73 (q,1H,J = 8.0 Hz),3.59 (m,1H),2.31-2.00 (m,4H),1.43 (s,9H),1.34 (d,J = 6.9 Hz,3H).
N-Boc-L-alanine-L-phenylalanine (Boc-Ala-Phe-OH): ESI-MS (m/z): 337.2 [M + H]+; 281.1 (M-(CH3)2C = CH2). 1H NMR (400 MHz,CDCl3): δ 7.32-7.10 (m,5H),6.85 (d,1H,J = 7.5 Hz),5.36-5.06 (m,1H),4.82 (q,1H,J = 6.5 Hz),4.21 (s,1H),3.20 (dd,1H,J = 14.0,5.5 Hz),3.03 (dd,1H,J = 14.3,6.4 Hz),1.43 (s,9H),1.26 (s,3H).
N-Boc-L-leucine-L-phenylalanine (Boc-Leu-Phe-OH): ESI-MS (m/z): 379.2 [M + H]+; 323.2 (M-(CH3)2C = CH2). 1H NMR (400 MHz,CDCl3): δ 7.29-7.11 (m,5H),6.97-6.78 (m,1H),5.13 (d,1H,J = 8.8 Hz),4.92-4.73 (m,1H),4.21 (d,1H,J = 8.2 Hz),3.26- 3.10 (m,1H),2.98 (dd,1H,J = 13.9,6.4 Hz),1.59 (m,2H),1.44 (m,10H),0.89 (t,6H,J = 7.0 Hz).
N-Boc-L-isoleucine-L-valine (Boc-Ile-Val-OH): ESI-MS (m/z): 331.2 [M + H]+; 275.2 (M-(CH3)2C = CH2). 1H NMR (400 MHz,CDCl3): δ 6.87 (d,1H,J = 8.7 Hz),5.34 (d,1H,J = 9.0 Hz),4.62 (dd,1H,J = 8.6,4.8 Hz),3.97 (t,1H,J = 8.2 Hz),2.00 (m,2H),1.43 (s,9H),1.22 (m,2H),0.93 (m,12H).
3. Results and discussionIn order to avoid racemization under alkaline conditions for the deprotection step,all the N-terminus of NHS ester were protected by Boc group. Most N-Boc protected NHS ester could be purchased,those commercially unavailable N-Boc protected NHS esters were readily obtained by coupling the corresponding N-Boc protected amino acids with N-hydroxysuccinimide (NHS-OH) in the presence of N,N-dicyclohexylcarbodiimide (DCC) [14]. The resulting byproducts containing the dicyclohexyl urea could be removed by filtration though a short pad of silica gel. After concentration of the filtrate,pure active esters could be recrystallized from various solvent systems. The solid N-Boc protected NHS esters were stable at -4 ℃ for a long period of time.
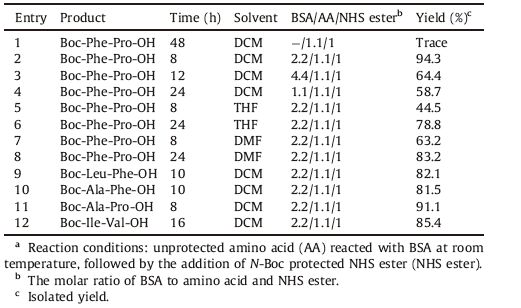
The coupling conditions and purification processes were optimized first through the synthesis of various dipeptides (Table 1). When no BSA was added,the coupling product was hardly detected (entry 1). Through screening various reaction conditions for the synthesis of Boc-Phe-Pro-OH,we found that the coupling efficiency and yield were optimal when 1.1 equiv. of unprotected proline reacted with 2.2 equiv. of BSA first in dichloromethane (DCM) at room temperature,followed by the addition of 1 equiv. of N-Boc protected Phe NHS ester (enrty 2). The ratio of BSA was important that either excessive (entry 3) or insufficient (entry 4) BSA would reduce the coupling yield significantly. Meanwhile,the ratio of each reagent was important not only for coupling efficiency,but also for purification process. NHS esters were insoluble in water,but soluble in organic solvents. In contrary,unprotected amino acid and BSA were either soluble in water or easily hydrolyzed in water. Therefore,the molar quantity of the NHS ester should be slightly lower than the unprotected amino acid and BSA to guarantee the NHS ester to be exhausted completely. Then the excessive unprotected amino acids and BSA could be easily removed by simply washing with water.
|
|
Table 1 Synthesis of N-Boc protected dipeptides via the BSA/NHS methoda. |
{kind=link}
All the unprotected amino acids are insoluble in organic solvents. So the addition order of reagents is quite important and unprotected amino acids should react with BSA first to increase its solubility and nucleophilicity. According to our data,the solubility of most amino acids improved significantly after being silylated with BSA. Among them,the silylation of proline was the fastest and it became soluble in dichloromethane after just 1 h reaction with BSA. But it took hours for other unprotected amino acids to be silylated and dissolve in dichloromethane. Therefore,the slightly lower yield when C-terminal was unprotected amino acids other than proline may be owing to their relatively poorer solubility. For the same reason,when C-terminal was several unprotected hydrophilic amino acids,such as aspartic acid,glutamic acid and cysteine,nearly no dipeptide products were obtained. Besides,unprotected basic amino acids that have two amino groups,such as arginine,lysine and histidine,are also unsuitable for the BSA/NHS method. The solubility of silylated amino acid in THF and DMF was clearly worse and even proline could not totally dissolve after reacting with BSA for 24 h. Consequently,the coupling efficiency was lower when the solvent was changed to THF (entry 5) or DMF (entry 7),and the time for achieving an acceptable yield in this two solvents was longer too (entries 6 and 8).
Excessive addition of BSA would silylated the carboxyl group and amino group simultaneously. When the carboxyl group and amino group were both silylated,it has been proved that acylating agents reacted with N-trimethylsilyl group exclusively [15]. The selectivity may be owing to the stability of silicon-oxygen bond is higher than that of silcon-nitrogen bond and silylation could increase the nucleophilicity of amines. Therefore,NHS esters react with amines selectively under these mild conditions (Scheme 1).
After all active ester being consumed as judged by TLC analysis,the dipeptide product was isolated through a convenient purification process. The silicon-oxygen bond could be easily hydrolyzed by water to produce the targeted N-Boc protected dipeptide [15d,e]. Besides,excessive BSA and amino acids are either hydrolysable or water soluble and the water-insoluble NHS esters are exhausted,all the excessive reagents and byproducts could be removed simply by water or saturated sodium chloride solution wash. The coupling reaction and purification process were both performed under mild conditions and no additional coupling reagents or acid/base were involved. Consequently,racemization was minimal. As expected,none of excessive reactants,byproducts or the racemized products was detected by HPLC analysis (Fig. 2).

|
Download:
|
| Fig. 2.HPLC analysis of Boc-Phe-Pro-OH synthesized using the BSA/NHS strategy. Chromatographic conditions: Instrument: Waters Acquity UPLC; Chromatographic column: Waters UPLC BEH C18 reversed-phase column (1.7 mm,50 mm × 2.1 mm); Flow rate: 0.2 mL/min; Column temperature: 40 ℃; Mobile phase: phase A: 0.1% formic acid aqueous solution; phase B: CH3CN; gradient conditions: 0 min!6 min: 95% phase A!5% phase A; 6 min!8 min: 5% phase A; 8 min!8.1 min: 5% phase A!95% phase A; 8.1 min!10 min: 95% phase A!5% phase A. | |
{kind=link}
Then the coupling and purification method was verified through the synthesis of another four dipeptides,all of which were obtained in high isolated yield of above 80 percent (entries 9-12). The N-Boc protecting group was subsequently cleaved using trifluoroacetic acid/dichloromethane (1:2) and the pure deprotected dipeptide was obtained after additional recrystallization from diethyl ether. More impurities could be observed when pure trifluoroacetic acid was utilized as the deprotection reagent,leading to lower yields and more complicated purification processes. When the dipeptides were produced in the form of hydrochloride salts,they would be more hygroscopic. Above all,we could synthesize the dipeptide products in good yields and high purity in significantly shorter reaction time with simpler purification processes.
4. ConclusionIn summary,the BSA/NHS strategy has been successfully utilized in the rapid,large scale solution-phase synthesis of various dipeptides. Through the BSA/NHS strategy,coupling reaction was completed under neutral and mild conditions that involve no extra coupling reagents or acid/base. Excessive reagents and byproducts were removed using water or saturated sodium chloride solution rather than several rounds of acidic and basic aqueous extractions. Moreover,all the reactants are inexpensive and widely used in conventional drug production. Above all,the BSA/NHS strategy has the potential to be applied in further commercial-scale manufacture of other peptide drugs.
AcknowledgmentThis research was financially supported by the National Science and Technology Major Project of China (No. 521042).
| [1] | V. du Vigneaud, C. Ressler, J.M. Swan, et al., The synthesis of an octapeptide amide with the hormonal activity of oxytocin, J. Am. Chem. Soc. 75(1953) 4879-4880. |
| [2] | C.M. Cloutheir, J.N. Pelleteir, Expanding the organic toolbox:a guide to integrating biocatalysis in synthesis, Chem. Soc. Rev. 41(2012) 1585-1605. |
| [3] | B.L. Bray, Large-scale manufacture of peptide therapeutics by chemical synthesis, Nat. Rev. Drug Discov. 2(2003) 587-593. |
| [4] | S. Maher, B. Ryan, A. Duffy, et al., Formulation strategies to improve oral peptide delivery, Pharm. Pat. Anal. 3(2014) 313-336. |
| [5] | (a) T.R. Pearce, K. Shroff, E. Kokkoli, Peptide targeted lipid nanoparticles for anticancer drug delivery, Adv. Mater. 24(2014) 3803-3822;(b) L.T. Eliassen, G. Berge, B. Sveinbjornsson, et al., Evidence for a direct antitumor mechanism of action of bovine lactoferricin, Anticancer Res. 22(2002) 2703-2710. |
| [6] | (a) L.L. Baggio, Q. Huang, T.J. Brown, et al., A recombinant human glucagon-like peptide (GLP)-1-albumin protein (Albugon) mimics peptidergic activation of GLP-1 receptor-dependent pathways coupled with satiety, gastrointestinal motility, and glucose homeostasis, Diabetes 53(2004) 2492-2500;(b) Q. Xiao, J. Giguere, M. Parisien, et al., Biological activities of glucagon-like peptide-1 analogues in vitro and in vivo, Biochemistry 40(2001) 2860-2869. |
| [7] | C.D. Fiell, J.A. Hiss, R.E.W. Hancock, et al., Designing antimicrobial peptides:form follows function, Nat. Rev. Drug Discov. 11(2012) 37-51. |
| [8] | S. Matsunaga, N. Fusetani, K. Hashimoto, et al.,F. Theonellamide, A novel antifungal bicyclic peptide from a marine sponge Theonella sp, J. Am. Chem. Soc. 111(1989) 2582-2588. |
| [9] | (a) N. Kaur, X. Lu, M.C. Gerhengorn, et al., Thyrotropin-releasing hormone (TRH) analogues that exhibit selectivity to TRH receptor subtype 2, J. Med. Chem. 48(2005) 6162-6165;(b) J. Rivier, W. Vale, M. Monahan, et al., Synthetic thyrotropin-releasing factor analogs. 3. Effect of replacement or modification of histidine residue on biological activity, J. Med. Chem. 15(1972) 479-482. |
| [10] | R.B. Merrifield, Solid phase peptide synthesis. (Ⅰ). The synthesis of a tetrapeptide, J. Am. Chem. Soc. 85(1963) 2149-2154. |
| [11] | T. Bruckdorfer, O. Marder, F. Albericio, From production of peptides in milligram amounts for research to multi-tons quantities for drugs of the future, Curr. Pharm. Biotechnol. 5(2004) 29-43. |
| [12] | (a) E. Bayer, M. Mutter, Liquid phase synthesis of peptides, Nature 237(1972) 512-513;(b) J. Wu, G. An, S. Lin, et al., Solution-phase-peptide synthesis via the groupassisted purification (GAP) chemistry without using chromatography and recrystallization, Chem. Commun. 50(2014) 1259-1261;(c) G. Tana, S. Kitada, S. Fujita, et al., A practical solution-phase synthesis of an antagonistic peptide of TNF-abased on hydrophobic tag strategy, Chem. Commun. 46(2010) 8219-8221;(d) M. Mizuno, K. Goto, T. Miura, et al., A novel peptide synthesis using fluorous chemistry, Chem. Commun. 8(2003) 972-973;(e) D. Takahashi, T. Yano, T. Fukui, Novel diphenylmethyl-derived amide protecting group for efficient liquid-phase peptide synthesis:AJ(Ⅰ)PHASE, Org. Lett. 14(2014) 4514-4517;(f) N. Naganna, N. Madhavan, Soluble non-cross-linked poly(norbornene) supports for peptide synthesis with minimal reagents, J. Org. Chem. 79(2014) 11549-11557;(g) Y. Fujita, S. Fujita, Y. Okada, et al., Soluble tag-assisted peptide head-to-tail cyclization:total synthesis of mahafacyclin B, Org. Lett. 15(2013) 1155-1157. |
| [13] | ChristianA.G.N. Montalbetti, V. Falque, Amide bond formation and peptide coupling, Tetrahedron 61(2005) 10827-10852. |
| [14] | M. Mikolajczyk, P. Kielbasinski, Recent developments in the carbodiimide chemistry, Tetrahedron 37(1981) 233-284. |
| [15] | (a) J.F. Klebe, in:E.C. Taylor (Ed.), Advances in Organic Chemistry, vol. 8, John Wiley and Sons, (Ⅰ)nc, New York, NY, 1972, p. 124;(b) B. Rigo, C. Lespagnol, M. Pauly, Studies on pyrrolidinones. Synthesis of Nacylpyroglutamic esters with bactericide and fungicide properties, J. Heterocycl. Chem. 25(1988) 49-57;(c) C. Bolm, A. Kasyan, K. Drauz, et al., α-Trialkylsilyl-substituted α-amino acid, Angew. Chem. (Ⅰ)nt. Ed. Engl. 39(2000) 2288-2290;(d) A.B. Ouryupina, V.Y. Komissarova, P.V. Petrovskiia, et al., Synthesis of n-phosphorylated aminoacids, Phosphorus Sulfur Silicon Relat. Elem. 103(1995) 215-224;(e) H. Fu, Z.L. Li, Y.F. Zhao, et al., Oligomerization of n,o-bis(trimethylsilyl)-α-amino acids into peptides mediated by o-phenylene ohosphorochloridate, J. Am. Chem. Soc. 121(1999) 291-295;(f) R.P. Singh, T. Umemoto, 4-Fluoropyrrolidine-2-carbonyl fluorides:useful synthons and their facile preparation with 4-tertbutyl-2,6-dimethylphenylsulfur trifluoride, J. Org. Chem. 76(2011) 3113-3121. |