 2016, Vol.27
2016, Vol.27 



b Department of Biotechnology, Savitribai Phule Pune University, Pune 411007, MS, India
Leishmaniasis is a family of parasitic diseases caused by infection of macrophages by obligate intracellular parasites of genus Leishmania. It is a major health problem worldwide and over 20 million people in 88 countries have one of the various forms of the disease [1]. Parasites transmissions occur by way of female sandflies via anthroponotic or zoonotic cycles [2]. Leishmaniasis is classified as cutaneous,visceral (Kala Azar),mucosal or mucocutaneous and diffused cutaneous on the basis of the evaluation of the patients and parasite [3]. Visceral leishmaniasis (VL or Kalaazar) is the most devastating form and arises from invasion of the reticuloendothelial system (spleen,liver and bone marrow) by the haemo flagellate protozoan parasite Leishmania donovani (L. donovani) [4]. While several drug treatment strategies are known,these drugs have the drawbacks of high expense,requirement of prolonged treatment,high toxicity to liver and heart,and the development of clinical resistance after a few weeks of treatment. These issues contribute to poor treatment outcomes and to an increase in leishmaniasis-AIDS co-infections in some countries [5]. Therefore,there is an urgent need for development of new,inexpensive,effective and safe drugs having potential antileishmanial activity. Oxidative and free-radical-mediated processes reactions seem to play an important role in the progression of various diseases,including cancer and coronary heart disease [6]. Radical damage has also been implicated in neurodegeneration occurring both through normal aging processes and in diseases such as Alzheimer’s disease [7]. Thus,there is also a need for development of safe and effective antioxidants to prevent radicalmediated damage.
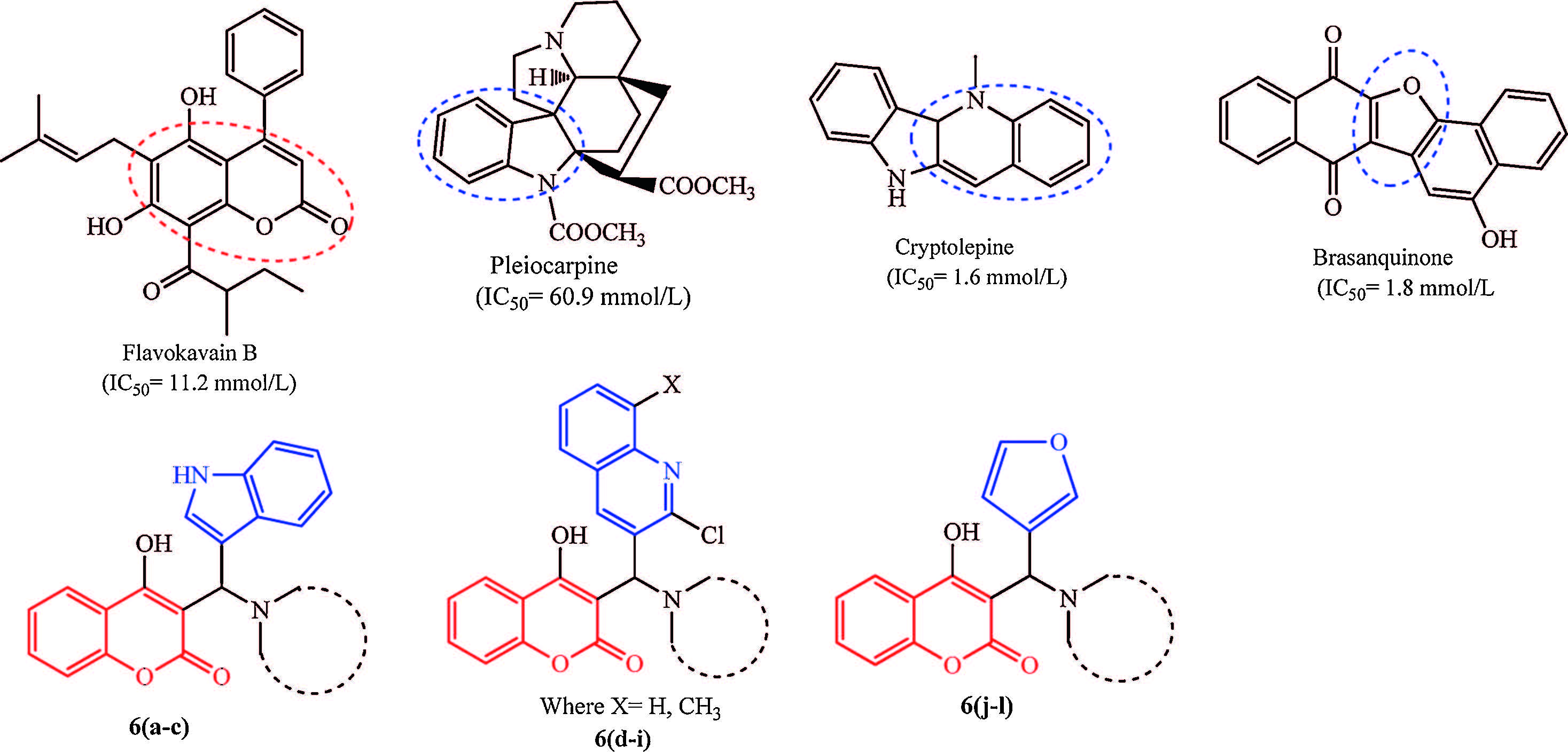
The coumarins are an important heterocyclic core structure that is present in numerous natural products with wide range of biological activities [8]. Flavokavain B,for example,is a natural coumarin derivative that is effective against the promastigote form of L. donovani parasites [9]. Also,many coumarins known to have antioxidant effect [10]. A number of other heterocyclic core structures,such as,indoles,quinolines,and furans,are also present in various biologically active natural products and drugs,including those with potent antileishmanial activity [11, 12, 13]. Pleiocarpine (an indole alkaloid) [14],cryptolepine (a quinoline alkaloid) [15],and brasanquinone (a furan derivative) [16] are specific examples of natural antileishmanial agents with activity against L. donovani parasites (Fig. 1).

|
Download:
|
| Fig. 1.Structure of some natural antileishmanial compounds active against L. donovani promastigotes and model structure of titled compounds 6(a-l). | |
Natural products constitute a large portion of marketed drugs,particularly in the area of infectious diseases,and often show remarkable potency,selectivity and drug-like properties [17]. However,the numbers of natural products are limited; hence incorporating either different natural products or drug fragments may provide millions of combinations which will be enriched in biological activity and less toxicity. Taking into account all of the aforementioned,and as a part of our ongoing effort towards identifying novel bioactive compounds [18, 19],we decided to explore ways of combining the coumarin nucleus with fragments present in other antileishmanial agents,heterocycles such as indoles,quinolines,and furans. We have synthesized a novel series of 3-substituted-4-hydroxycoumarin hybrids 6(a-l) and evaluated them as antileishmanial agents against L. donovani (Fig. 1). Coumarin such as isopimpinellin is reported to act as antileishmanial agents through the inhibition of enzyme Adenine phosphoribosyltransferase enzyme [20]. Accordingly,we opted to dock our synthesized compounds against the Adenine phosphor-ibosyltransferase of L. donovani to understand features of the molecules that may be responsible for their antileishmanial activity. Because coumarins have also found use as antioxidants,the synthesized compounds were also evaluated for antioxidant activity using DPPH-radical scavenging method.We have also used in silico method to predict ADME properties to suggest the suitability of any of the new compounds for further drug development,particularly with respect to activity.
2. Experimental 2.1. ChemistryAll reagents and solvents used were purchased from the Sigma and Avra synthesis. The completion of reaction was checked by ascending thin layer chromatography (TLC) on silica gel-G (Merck) coated aluminum plates,visualized by iodine vapor. Infrared (IR) spectra were recorded for the compounds on JASCO FTIR (PS 4000) using KBr pallet. 1H NMR and 13CNMR spectra were recorded using Brucker Avance II. Multiplicities are recorded as s (singlet),d (doublet),t (triplet),q (quartet),and m (multiplet). Mass spectra were taken with Micromass-QUATTRO-II of WATER mass spectrometer. Elemental analyses (C,H,and N) were undertaken with a Shimadzu’s FLASHEA112 analyzer and all analyses were consistent with theoretical values (within ±0.4%),unless indicated.
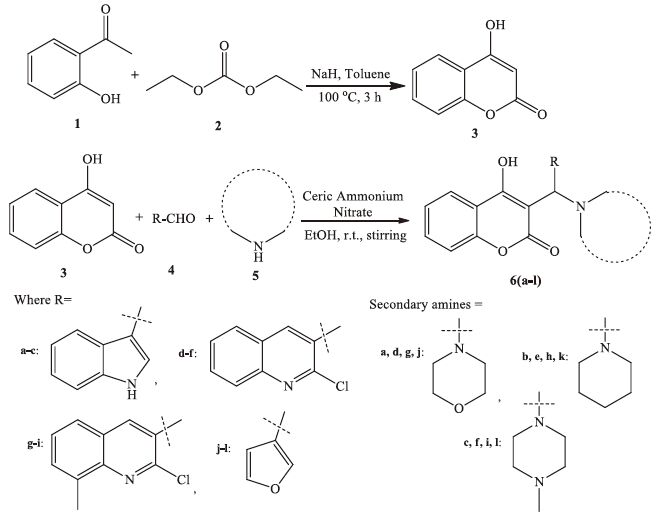
2.1.1. General procedure for synthesis of 3-substituted-4- hydroxycoumarin derivatives 6(a-l)A mixture of 4-hydroxycoumarin 3 (1.0 mmol),heterocyclic aldehydes 4 (1.0 mmol) and secondary amines 5 (1.0 mmol) was stirred at room temperature (25-30 ℃) in ethanol (15 mL) in the presence of catalytic amount of ceric ammonium nitrate (10 mol%) for 15-20 min. After completion of the reaction as monitored by TLC analysis,the reaction mixture was poured into water. The solid product formed was filtered,dried and recrystallized using ethanol.
2.2. Biological evaluations 2.2.1. In vitro antileishmanial activityThe assay for in vitro antileishmanial activity on culture of L. donovani promastigotes (NHOM/IN/80/DD8) was carried out in 96-well tissue culture plates using reported procedure [21]. The promastigotes culture was maintained at 22 ℃ inmodified RPMI 1640 pH 7.4 (without phenol red) with 10% FCS medium. Drug dilutions were prepared in DMSO and appropriate concentration of each drug was used in triplicate. Plates were incubated at 22 ℃ for 72 h and evaluated using modified MTT assay,where the conversion of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan by mitochondrial enzymes served as an indicator of cell viability and the amount of formazan produced was directly proportional to the number of metabolically active cells. Accordingly,absorbance at 492 nm represented the number of live cells. The concentration that decreased cell growth by 50% (IC50) was computed from growth inhibition curve. Pentamidine and miltefosine were used as standard drugs.
2.2.2. Antioxidant activity (DPPH radical scavenging method)The hydrogen atom or electron donation ability of the compounds was measured from the bleaching of the purple colored methanol solution of 1,1-diphenyl-1-picrylhydrazyl (DPPH) [22]. The spectrophotometric assay uses the stable radical DPPH as a reagent. 1 mL of various concentrations of the test compounds (5,10,25,50 and 100 μg/mL) in methanol was added to 4 mL of 0.004% (w/v) methanol solution of DPPH. After a 30 min incubation period at room temperature,the absorbance was read against blank at 517 nm. The natural antioxidant ascorbic acid and synthetic antioxidant BHT (butylated hydroxy toluene) were used as standards. The percent of inhibition (I%) of free radical production from DPPH was calculated by the following equation.
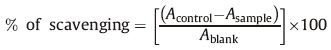
Cytotoxic study of the synthesized compounds on HeLa cell line was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method [23]. In brief,exponentially growing Hela cells (1 ± 104 cells/well) were seeded in a 96-well culture plate and incubated at 37 ℃ in a 5% CO2 incubator for 24 h. The drugs were dissolved in 0.1% DMSO and then diluted with the medium. The cells were then exposed to different concentrations of drug (0-10 μg/mL) and incubated for 72 h. The cells in the control wells received medium containing the same volume of DMSO (0.1%). After incubation,5 μg/mL of MTT reagent was added in each well and further incubated for additional 4 h. The formazan produced by the viable cells was stabilized by addition of 0.1 mL DMSO. The absorbance of each well at =0 nmwas determined by a microplate spectrophotometer. The cells were also seen under the microscope (Ziess,Germany) at 10× magnification.
2.3. Computational studies 2.3.1. Docking studyDocking study of synthesized compounds 6(a-l) was performed using VLife MDS 4.3 package [24]. With this purpose,crystal structure of Adenine phosphoribosyltransferase of L. donovani (PDB ID: 1QB8) [25] was obtained from the Protein Data Bank in order to prepare protein for docking study. The cavities in the receptor were mapped to assign an appropriate active site. The basic features used to map the cavities were the surface mapping of the protein and identifying the geometric voids as well as scaling the void for its hydrophobic characteristics using V Life MDS analyze tool. Hence all the cavities that are present in Adenine phosphoribosyl-transferase protein were identified and ranked based on their size and hydrophobic surface area. Considering the dimensions and hydrophobic surface area,ranked-1 cavity was chosen for docking study.
2.3.2. ADME predictionA computational study of synthesized compounds 6(a-l) was performed for prediction of ADME properties. In this study,we calculated molecular volume (MV),molecular weight (MW),logarithm of partition coefficient (miLog P),number of hydrogen bond acceptors (n-ON),number of hydrogen bonds donors (n- OHNH),topological polar surface area (TPSA),number of rotatable bonds (n-ROTB) and Lipinski’s rule of five [26] using Molinspiration online property calculation toolkit [27]. Absorption (% ABS) was calculated by: %ABS = 109 × (0.345 ± TPSA) [28].
3. Results and discussion 3.1. ChemistryThe starting material 4-hydroxycoumarin (3) was synthesized using 2-hydroxyacetophenone (1) and diethyl carbonate (2) as per reported method [29]. Then a one-pot three component reaction [30] was used to synthesize 3-substituted-4-hydroxycoumarin derivatives 6(a-l) as shown in Scheme 1. These products were obtained via the reaction of 4-hydroxycoumarin (3) with various heterocyclic aldehydes (4) and secondary amines (5) in ethanol at room temperature,using ceric ammonium nitrate (CAN,10 mol%) as catalyst,through a Mannich type reaction mechanism. CAN has received considerable attention as an relatively inexpensive,nontoxic,water soluble and easily handled catalyst for the construction of carbon-carbon and carbon-heteroatom bonds [31]. The formation of the desired compound indicates that CAN play an important role for the rapid formation of the imine intermediate. The catalyst may induce 4-hydroxy coumarin to act as the Mannich donor for the very fast formation of 3-substituted-4-hydroxycoumarin derivatives. The rapid imine generation and subsequent ‘C- C’ bond formation within a very short time catalyzed by CAN are the attractive features of this protocol.

|
Download:
|
| Scheme 1.Synthetic protocol for titled compounds 6(a-l). | |
To optimize the reaction conditions,we studied a model reaction of 4-hydroxycoumarin (1.0 mmol),indole-3-carbaldehyde (1.0 mmol) and morpholine (1.0 mmol) in ethanol to synthesize compound 6a. A wide variety of catalysts were surveyed,including oxalic acid,boric acid,sulphamic acid,p-toluenesulphonic acid,ZnO,ZnCl2,and ceric ammonium nitrate (CAN),all at 30 mol%. CAN as the catalyst gave the best results in terms of yield (98%) and reaction time (15 min) (Table S1 in Supporting information). Solvent effects were then studied,using water,toluene,dimethylsulfoxide,ethanol,and dichloromethane. Ethanol was found to be best among the studied solvents (Table S2 in Supporting information). For the optimization of catalyst loading,we used CAN as catalyst at 0,5,10,15,20,and 30 mol%. The results (Table S3 in Supporting information) revealed that 10 mol% CAN is fully sufficient for obtaining a high yield. This low catalyst loading contributes to the efficiency and simplicity of the process.
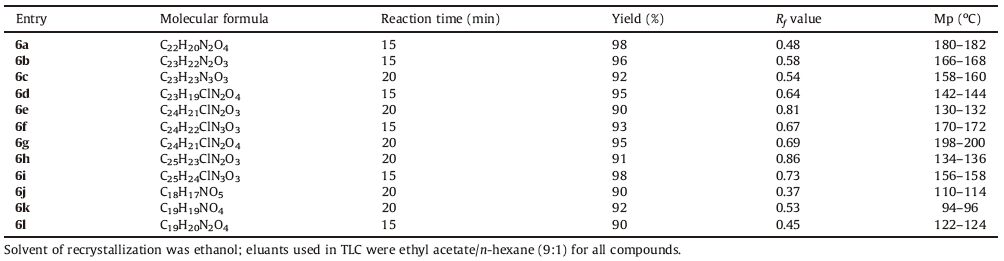
After optimization of the model reaction,the scope of the transformation was then studied. We further expanded the series and synthesized 12 novel derivatives of 3-substituted-4-hydroxycoumarins 6(a-l) by reacting 4-hydroxycoumarin 3,heterocyclic aldehydes 4,and secondary amines 5 in ethanol using CAN (10 mol%) as catalyst at room temperature (25-30 ℃). The reaction preceded smoothly under mild reaction conditions. The isolated yield of synthesized compounds was in the range of 90-98% and reactions were completed in 15-20 min (monitored by TLC). The work-up of these reactions was simple,required only a filtration. The compounds were thus obtained in pure form without aids of any system like chromatography. Melting points were determined in open capillary tubes and are uncorrected. The physical data for the compounds 6(a-l) are presented in Table 1,with characterization by IR,1H NMR,13C NMR,Mass and elemental spectroscopic data,with results in agreement with the proposed structures (Supporting information). This type of Mannish adducts are prone to retro-Mannich decomposition to lose the amine and form a Michael acceptor [32]. In order to check the thermal stability of the synthesized compounds 6(a-l),we subjected compound 6i for stress stability testing as per ICHQ1A 2.1.2 guidelines [33]. Compound 6i was subjected to forced degradation study at temperatures 105 ℃ and 157 ℃ for 1 month. After 1 month of study,compound 6i was again characterized for physical and for spectroscopic data. The data suggested that compound 6i was stable even after 1 month of force degradation study (stress study).
|
|
Table 1 Physical data for 3-substituted-4-hydroxycoumarin derivatives 6(a-l). |
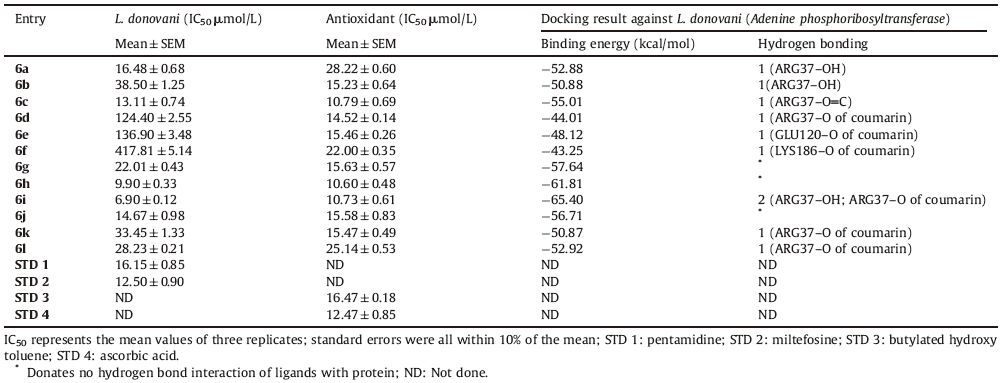
The title compounds 6(a-l) were tested in vitro for their antileishmanial activity against a culture of L. donovani promastigotes and data obtained is depicted in Table 2. Most of the synthesized compounds showed promising antileishma-nial activity. Compounds 6a,6c,6h,6i,and 6j (IC50 values = 6.90-16.48 μmol/L) were most promising,with activity comparable to that of standard drugs. Compounds 6b,6g,6k,and 6l were less potent in this assay (IC50 values = 22.01- 38.50 μmol/L). The compounds 6d,6e,and 6f (R = 2-chloroquinolinyl) were found to inactive against culture of L. donovani promastigotes. The compounds 6h (IC50 = 9.90 μmol/L) and 6i (IC50 = 6.90 μmol/L) were more potent than standard drugs pentamidine ((IC50 = 16.15 μmol/L) and miltefosine (IC50 = 12.5 μmol/L).
|
|
Table 2 Biological evaluation and molecular docking statistics of synthesized compounds 6(a-l). |
To establish structure-activity relationship (SAR) in the series,we divided the synthesized compounds into three classes,the indole class 6(a-c),quinoline class 6(d-i),and furan class 6(j- l). The activity mainly depends upon the presence of the different heterocycles and secondary amines at the 3rd position of the 4- hydroxycoumarin scaffold. The indole class showed promising activity,particularly when a 4-methylpiperazine group was present at 3rd position of 4-hydroxycoumarin (6c). From the quinolines 6(d-i),a 2-chloroquinolinyl group at 3rd position of 4- hydroxycoumarin (compounds 6d,6e,and 6f) provides much lower activity towards L. donovani promastigote cultures. Replacement of the 2-chloroquinolinyl group with 2-chloro-8-methylquinolinyl (compounds 6g,6h,and 6i) increased the activity by 5-70 folds. Adding a methyl group at the 8th position of quinoline further enhanced activity. Substitution of 2-chloro-8-methylquinolinyl in combination with a 4-methylpiperazinyl group at 3rd position of the 4-hydroxycoumarin core led to the most potent compound of the series (6i). From the furan class 6(j-l),the furanyl nucleus in combination with morpholine group (compound 6j) at 3rd position of the 4-hydroxycoumarin gave the good antileishmanial activity.
3.2.2. Antioxidant activity (DPPH radical scavenging method)Antioxidant activity was assessed in vitro by using the DPPH radical scavenging assay method. The DPPH has an odd electron so it can accept an electron or hydrogen free radical. In the presence of antioxidant,this odd electron becomes paired due to H transfer from antioxidant and hence DPPH absorbance decreases. All the synthesized compounds 6(a-l) showed interesting antioxidant activity (IC50 = 10.60-28.22 μmol/L) when compared to the standards (Table 2). The common antioxidant activity in the series is likely due to the presence of electron-releasing hydroxy at the 4th position of coumarin nucleus. All the compounds except 6a (IC50 = 28.22 μmol/L),6f (IC50 = 22.00 μmol/L),and 6l (IC50 = 25.14 μmol/L) were more active than standard butylated hydroxy toluene (IC50 = 16.47 μmol/L). The most potent antioxidants in the series were 6c (IC50 = 10.79 μmol/L),6h (IC50 = 10.60 μmol/L),and 6i (IC50 = 10.73 μmol/L),comparing favorably to the standards BHT (IC50 = 16.47 μmol/L) and ascorbic acid (IC50 = 12.69 μmol/L). Among the synthesized compounds,quinoline series 6(d-i) had shown better activity than indole 6(a- c) and furan 6(j-l) series. The higher activity for quinolines may arise due to the presence of an imine unit in the quinoline moiety,which can stabilize an adjacent radical [34]. The higher radical scavenging activity for compounds 6g,6h,and 6i (R = 2-chloro-8- methylquinol-3-yl) can also be explained by noting that the methyl group also helps stabilize an adjacent unpaired electron [35].
3.2.3. In vitro cytotoxicity studyTo test for cytotoxicity of titled compounds,we exposed HeLa cells to the test compounds 6(a-l) and observed the cell morphology. None of the synthesized compounds showed cytotoxicity towards the HeLa cell line up to highest tested concentrations,suggesting the potential for low intrinsic toxicity toward human cells. The lack of cytotoxicity effects of compound 6i on the HeLa cell line is shown in Fig. 2.

|
Download:
|
| Fig. 2.Cytotoxic study of compound 6i. | |
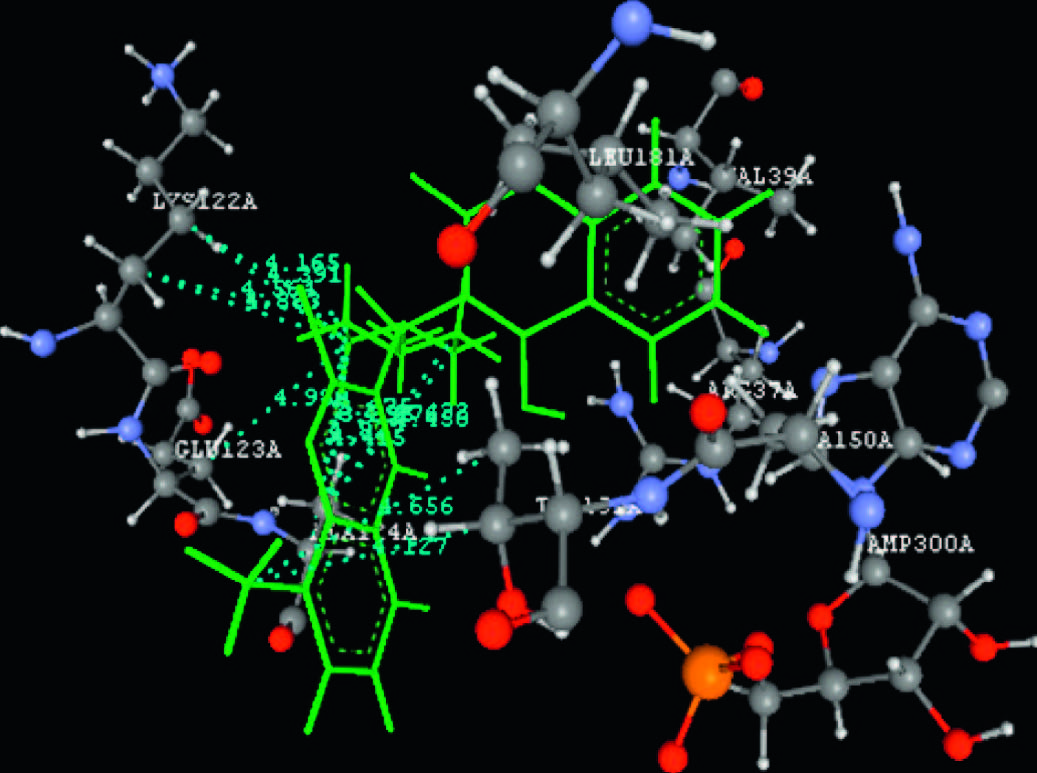
A molecular docking study of the synthesized compounds 6(a- l) was performed against the Adenine phosphoribosyltransferase enzyme of L. donovani to understand the possible binding mode for the test compounds. Adenine phosphoribosyltransferase,a Leishmania enzyme,is responsible for pyrophosphorolysis in sequencing and amplification of nucleic acids in protein synthesis [36] and is a potential target for chemotherapeutic intervention. The docking calculations and hydrogen bonds interactions noted are shown in Table 2. The modeling results for compounds 6(a-l) correlate in general with the observed antileishmanial activity. The docking results indicated that the 3-substituted-4-hydroxycoumarin core of the compounds 6(a-l) is held in the active site by a combination of various hydrophobic and van der Waals interactions with the enzyme. Major hydrophobic interactions occur between the 3-substituted-4-hydroxycoumarin core and with the active side chain of VAL39,PRO40,GLU120,TYR121,LYS122,GLU123,ALA124,ALA150,THR151,and LEU181. The various methylene groups (-CH2-) form strong hydrophobic interactions with active site hydrophobic residues. H-bonding groups in the coumarin core (OH,C=O,and -O-) formed hydrogen bonds with ARG37,GLU120,and LYS186,thus suggesting that the central core is important for inhibiting of Adenine phosphoribosyltransferase enzyme of L. donovani.
The most active antileishmanial compound 6i showed the lowest binding energy in the docking study,-65.40 kcal/mol. The interactions of compound 6i with enzyme are shown in Fig. 3. The amino acid ARG37 (2.589Å ) forms hydrogen bonds with the -OH and -O- groups of the 4-hydroxycoumarin nucleus of compound 6i. Also,both methyl groups of compound 6i fit well into the active site,forming hydrophobic interactions with the THR151 amino residue. This observation suggests that a 2-chloro-8-methylquinolinyl moiety in combination with 4-methylpiperazinyl group at 3rd position of 4-hydroxycoumarin ring has optimal interactions with the Adenine phosphoribosyltransferase enzyme.

|
Download:
|
| Fig. 3.Docking study of compound 6i with Adenine phosphoribosyltransferase of L.donovani (PDB ID: 1QB8). Ligands are shown in green color. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.) | |
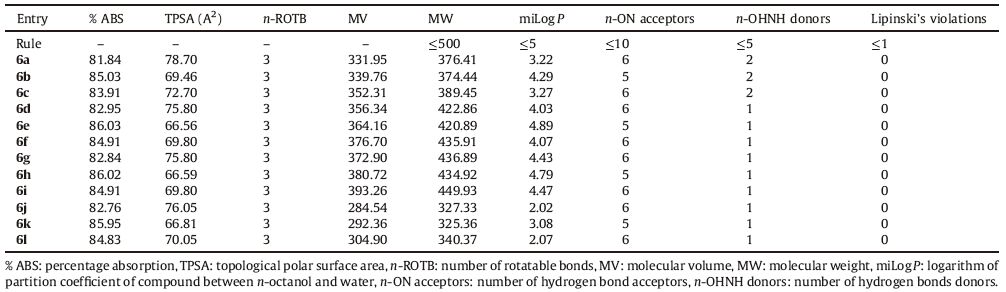
Issues of druggability are also important considerations. As shown in Table 3,all of the synthesized compounds exhibited a good % ABS (81.84-86.03%). Furthermore,none of the compounds violated Lipinski’s rule of five,suggesting promise for development as good drug candidates. A molecule likely to be developed as an orally active drug candidate should show no more than one violation of the following four criteria: log P (octanol-water partition coefficient) ≤ 5,molecular weight ≤ 500,number of hydrogen bond acceptors ≤ 10 and number of hydrogen bond donors ≤ 5 [37]. All the synthesized compounds 6(a-l) followed the criteria for orally active drug and therefore,these compounds can be further developed as oral drug candidates.
|
|
Table 3 Pharmacokinetic parameters important for good oral bioavailability of synthesized compounds 6(a-l). |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In conclusion,we have successfully designed compounds 6(a-l) based on a natural product-inspired scaffolds. Also,we have developed an expeditious one-pot three component method for the synthesis of novel 3-substituted-4-hydroxycoumarin derivatives 6(a-l) from 4-hydroxycoumarin,heterocyclic aldehydes and secondary amines in ethanol at room temperature using CAN (10 mol%) as catalyst. The advantages of this method include mild reaction conditions,simplicity,short reaction time,easy work up,and high yields. Several compounds have significant antileishmanial activity against L. donovani promastigotes and many also have antioxidant activity. The compounds 6h (IC50 = 9.90 μmol/L) and 6i (IC50 = 6.90 μmol/L) from the quinoline class were the most active antileishmanial agents. The compound 6i with 2-chloro-8- methylquinolinyl in combination with 4-methylpiperazinyl group at 3rd position of 4-hydroxycoumarin was most potent antileishmanial agent in the series. Three compounds 6c (IC50 = 10.79 mmol/ L),6h (IC50 = 10.60 μmol/L),and 6i (IC50 = 10.73 μmol/L) were shown potent antioxidants when compared with standards. None of the synthesized compounds were cytotoxicity to HeLa cell lines upto their highest tested concentrations. A molecular docking study suggested the binding interactions of these compounds with Adenine phosphoribosyltransferase of L. donovani. Furthermore,analysis of the ADME parameters for synthesized compounds suggested that they have good drug-like properties with potential for oral delivery. The lead compounds will be investigated further in an effort to develop more active,safe and cost-effective antileishmanial and antioxidant agents.
AcknowledgmentsThe authors are thankful to the Mrs. Fatma Rafiq Zakaria,Chairman,Maulana Azad Educational Trust and Principal,Y.B. Chavan College of Pharmacy,Dr. Rafiq Zakaria Campus,Aurangabad 431 001 (M.S.),India for providing the laboratory facility. The authors are also thankful to Bar C. (Department of Zoology,University of Pune) for providing L. donovani culture. The authors are also thankful to SAIF,Punjab University,Chandigarh,India for providing NMR spectra.
Appendix A. Supplementary dataSupplementary material related to this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.10. 028.
| [1] | (a) J.D. Berman, Human leishmaniasis: clinical, diagnostic, and chemotherapeutic developments in the last 10 years, Clin. Infect Dis. 24 (1997) 684-703;(b) M. Khaw, C.B. Panosian, Human antiprotozoal therapy: past, present, and future, Clin. Microbiol. Rev. 8 (1995) 427-439. |
| [2] | R. Reithinger, J.C. Dujardin, H. Louzir, et al., Cutaneous leishmaniasis, Lancet Infect. Dis. 7 (2007) 581-596. |
| [3] | World Health Organization (WHO), Tropical Disease Research Progress, World Health Organization (WHO), 2001. |
| [4] | R.W. Ashford, P. Desjeux, P. DeRaadt, Estimation of population at risk of infection and number of cases of leishmaniasis, Parasitol. Today 8 (1992) 104-105. |
| [5] | (a) H.W. Murray, Treatment of visceral leishmaniasis in 2004, Am. J. Trop. Med. Hyg. 71 (2004) 787-794;(b) S.L. Croft, Recent developments in the chemotherapy of leishmaniasis, Trends Pharmacol. Sci. 9 (1988) 376-381;(c) J.D. Berman, Chemotherapy for leishmaniasis: biochemical mechanisms, clinical efficacy, and future strategies, Rev. Infect Dis. 10 (1988) 560-586. |
| [6] | K.F. Gey, The antioxidant hypothesis of cardiovascular disease: epidemiology and mechanisms, Biochem. Soc. Trans. 18 (1990) 1041-1045. |
| [7] | (a) M.A. Smith, G. Perry, P.L. Richey, et al., Oxidative damage in Alzheimer's, Nature 382 (1996) 120-121;(b) M.N. Diaz, B. Frei, J.A. Vita, J.F. Keaney, Antioxidants and atherosclerotic heart disease, N. Engl. J. Med. 337 (1997) 408-416. |
| [8] | R.M. Wilson, S.J. Danishefsky, Small molecule natural products in the discovery of therapeutic agents: the synthesis connection, J. Org. Chem. 71 (2006) 8329-8351. |
| [9] | M.J. Chan-Bacab, L.M. Peña-Rodríguez, Plant natural products with leishmanicidal activity, Nat. Prod. Rep. 18 (2001) 674-688. |
| [10] | (a) I. Kostova, S. Bhatia, P. Grigorov, et al., Coumarins as antioxidants, Curr. Med. Chem. 18 (2011) 3929-3951;(b) I. Kostova, Synthetic and natural coumarins as antioxidants, Mini Rev. Med. Chem. 6 (2006) 365-374. |
| [11] | (a) L. Gupta, A. Talwar, Nishi, et al., Synthesis of marine alkaloid: 8,9-dihydrocoscinamide B and its analogues as novel class of antileishmanial agents, Bioorg. Med. Chem. Lett. 17 (2007) 4075-4079;(b) S.S. Chauhan, L. Gupta, M. Mittal, et al., Synthesis and biological evaluation of indolyl glyoxylamides as a new class of antileishmanial agents, Bioorg. Med. Chem. Lett. 20 (2010) 6191-6194. |
| [12] | (a) N.P. Sahu, C. Pal, N.B. Mandal, et al., Synthesis of a novel quinoline derivative, 2-(2-methylquinolin-4-ylamino)-N-phenylacetamide—a potential antileishmanial agent, Bioorg. Med. Chem. 10 (2002) 1687-1693;(b) Z. Dardari, M. Lemrani, A. Bahloul, et al., Antileishmanial activity of a new 8-hydroxyquinoline derivative designed 7-[5'-(3'-phenylisoxazolino)methyl]-8-hydroxyquinoline: preliminary study, Farmaco 59 (2004) 195-199. |
| [13] | (a) A. Tahghighi, S. Emami, S. Razmi, et al., New 5-(nitroheteroaryl)-1,3, 4-thiadiazols containing acyclic amines at C-2: synthesis and SAR study for their antileishmanial activity, J. Enzyme Inhib. Med. Chem. 28 (2013) 843-852;(b) C.S. Reid, A.F. Farahat, X.H. Zhu, et al., Antileishmanial bis-arylimidamides: DB766 analogs modified in the linker region and bis-arylimidamide structure-activity relationships, Bioorg. Med. Chem. Lett. 22 (2012) 6806-6810. |
| [14] | V. Muñoz, C. Morretti, M. Sauvain, et al., Isolation of bis-indole alkaloids with antileishmanial and antibacterial activities from Perschiera van heurkii (syn. Tabernaemontana van heurkii), Planta Med. 60 (1994) 455-459. |
| [15] | (a) A.G. Tempone, A.C.M.P. da Silva, C.A. Brandt, et al., Synthesis and antileishmanial activities of novel 3-substituted quinolines, Antimicrob. Agents Chemother. 49 (2005) 1076-1080;(b) J.N. Sangshetti, F.A.K. Khan, A.A. Kulkarni, R. Arote, R.H. Patil, Antileishmanial drug discovery: comprehensive review of the last 10 years, RSC Adv. 5 (2015) 32376-32415. |
| [16] | V.K. Marrapu, M. Mittal, R. Shivahare, S. Gupta, K. Bhandari, Synthesis and evaluation of new furanyl and thiophenyl azoles as antileishmanial agents, Eur. J. Med. Chem. 46 (2011) 1694-1700. |
| [17] | O. Kayser, A.F. Kiderlen, H. Laatsch, S.L. Croft, In vitro leishmanicidal activity of monomeric and dimeric naphthoquinones, Acta Trop. 77 (2000) 307-314. |
| [18] | (a) J.N. Sangshetti, D.B. Shinde, Synthesis of some novel 3-(1-(1-substitutedpiperidin-4-yl)-1H-1,2, 3-triazol-4-yl)-5-substituted phenyl-1,2,4-oxadiazoles as antifungal agents, Eur. J. Med. Chem. 46 (2011) 1040-1044;(b) J.N. Sangshetti, R.R. Nagawade, D.B. Shinde, Synthesis of novel 3-(1-(1-substituted piperidin-4-yl)-1H-1,2, 3-triazol-4-yl)-1, 2, 4-oxadiazol-5(4H)-one as antifungal agents, Bioorg. Med. Chem. Lett. 19 (2009) 3564-3567;(c) J.N. Sangshetti, D.B. Shinde, One pot synthesis and SAR of some novel 3-substituted 5,6-diphenyl-1, 2, 4-triazines as antifungal agents, Bioorg. Med. Chem. Lett. 20 (2010) 742-745;(d) J.N. Sangshetti, P.P. Dharmadhikari, R.S. Chouthe, et al., Microwave assisted nano (ZnO-TiO2) catalyzed synthesis of some new 4,5,6,7-tetrahydro-6-((5-substituted-1, 3,4-oxadiazol-2-yl)methyl)thieno[2,3-c] pyridine as antimicrobial agents, Bioorg. Med. Chem. Lett. 23 (2013) 2250-2253;(e) Z. Zaheer, F.A.K. Khan, J.N. Sangshetti, R.H. Patil, Efficient one-pot synthesis, molecular docking and in silico ADME prediction of bis-(4-hydroxycoumarin-3-yl) methane derivatives as antileishmanial agents, EXCLI J. 14 (2015) 935-947. |
| [19] | (a) J.N. Sangshetti, A.R. Chabukswar, D.B. Shinde, Microwave assisted one pot synthesis of some novel 2,5-disubstituted 1,3,4-oxadiazoles as antifungal agents, Bioorg. Med. Chem. Lett. 21 (2011) 444-448;(b) J.N. Sangshetti, R.I. Shaikh, F.A.K. Khan, et al., Synthesis, antileishmanial activity and docking study of N'-substitutedbenzylidene-2-(6,7-dihydrothieno [3,2-c] pyridin-5(4H)-yl)acetohydrazides, Bioorg. Med. Chem. Lett. 24 (2014) 1605-1610;(c) J.N. Sangshetti, F.A.K. Khan, R.S. Chouthe, M.G. Damale, D.B. Shinde, Synthesis, docking and ADMET prediction of novel 5-((5-substituted-1-H-1,2,4-triazol-3-yl) methyl)-4,5, 6,7-tetrahydrothieno[3,2-c] pyridine as antifungal agents, Chin. Chem. Lett. 25 (2014) 1033-1038;(d) J.N. Sangshetti, F.A.K. Khan, R.H. Patil, et al., Biofilm inhibition of linezolid-like Schiff bases: synthesis, biological activity, molecular docking and in silico ADME prediction, Bioorg. Med. Chem. Lett. 25 (2015) 874-880;(e) F.A.K. Khan, J.N. Sangshetti, Design, synthesis and molecular docking study of hybrid quinoline-4-YL-oxadiazoles/oxathiadiazoles as potent antifungal agents, Int. J. Pharm. Pharm. Sci. 7 (2015) 223-229. |
| [20] | M. Silva, H.B. Napolitano, J. Ellena, et al., 3-(5,7-Dimethoxy-2,2-dimethyl-2Hbenzo[b] pyran-6-yl) propionic acid: a potential inhibitor against Leishmania, Acta Cryst. E59 (2003) o1575-o1577. |
| [21] | A. Dutta, S. Bandyopadhyay, C. Mandal, M. Chatterjee, Development of a modified MTT assay for screening antimonial resistant field isolates of Indian visceral leishmaniasis, Parasitol. Int. 54 (2005) 119-122. |
| [22] | M. Burits, F. Bucar, Antioxidant activity of Nigella sativa essential oil, Phytother. Res. 14 (2000) 323-328. |
| [23] | F. Denizlt, R.T. Lang, Rapid colorimetric assay for cell growth and survival: modifications to the tetrazolium dye procedure giving improved sensitivity and reliability, J. Immunol. Methods 89 (1986) 271-277. |
| [24] | VLife, Molecular Design Suite 4.3, VLife Sciences Technologies Pvt. Ltd, 2015 hwww.Vlifesciences.comi. |
| [25] | C.L. Phillips, B. Ullman, R.G. Brennan, C.P. Hill, Crystal structures of adenine phosphoribosyltransferase from Leishmania donovani, EMBO J. 18 (1999) 3533-3545. |
| [26] | C.A. Lipinski, L. Lombardo, B.W. Dominy, P.J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev. 46 (2001) 3-26. |
| [27] | Molinspiration Chemoinformatics Brastislava, Slovak Republic, 2015 Available from hhttp://www.molinspiration.com/cgi-bin/propertiesi. |
| [28] | Y.H. Zhao, M.H. Abraham, J. Le, et al., Rate-limited steps of human oral absorption and QSAR studies, Pharm. Res. 19 (2002) 1446-1457. |
| [29] | J.C. Jung, Y.J. Jung, O.S. Park, A convenient one-pot synthesis of 4-hydroxycoumarin, 4-hydroxythiocoumarin, and 4-hydroxyquinolin-2(1H)-one, Synth. Commun. 31 (2001) 1195-1200. |
| [30] | (a) M. Mohsenimehr, M. Mamaghani, F. Shirini, M. Sheykhan, F.A. Moghaddam, One-pot synthesis of novel pyrido[2,3-d] pyrimidines using HAp-encapsulated-γ-Fe2O3 supported sulfonic acid nanocatalyst under solvent-free conditions, Chin. Chem. Lett. 25 (2014) 1387-1391;(b) J.N. Sangshetti, F.A.K. Khan, C.S. Kute, Z. Zaheer, R.Z. Ahmed, One-pot threecomponent synthesis of 3-(α-aminobenzyl)-4-hydroxycoumarin derivatives using nanocrystalline TiO2 as reusable catalyst, Russ. J. Org. Chem. 51 (2015) 69-73;(c) M.A. Ameen, S.M. Motamed, F.F. Abdel-latif, Highly efficient one-pot synthesis of dihydropyran heterocycles, Chin. Chem. Lett. 25 (2014) 212-214;(d) J.N. Sangshetti, F.A.K. Khan, R.S. Chouthe, Z. Zaheer, R.Z. Ahmed, Watermediated oxalic acid catalysed one-pot synthesis of 2-(substituted phenyl) phthalazin-1(2H)-ones, J. Taibah Univ. Sci. 9 (2015) 548-554. |
| [31] | (a) B. Han, X.D. Jia, X.L. Jin, et al., A CAN-initiated aza-Diels-Alder reaction for a facile synthesis of 4-amido-N-yl tetrahydroquinolines, Tetrahedron Lett. 47 (2006) 3545-3547;(b) J.N. Sangshetti, N.D. Kokare, S.A. Kotharkara, D.B. Shinde, Ceric ammonium nitrate catalysed three component one-pot efficient synthesis of 2,4,5-triaryl-1Himidazoles, J. Chem. Sci. 120 (2008) 463-467;(c) A.P.G. Nikalje, M.S. Ghodke, F.A.K. Khan, J.N. Sangshetti, CAN catalyzed onepot synthesis and docking study of some novel substituted imidazole coupled 1,2,4-triazole-5-carboxylic acids as antifungal agents, Chin. Chem. Lett. 26 (2015) 108-112. |
| [32] | B.D. Mather, K. Viswanathan, K.M. Miller, T.E. Long, Michael addition reactions in macromolecular design for emerging technologies, Prog. Polym. Sci. 31 (2006) 487-531. |
| [33] | (a) IFPMA, Stability testing of new drug substances and drug products ICH Q1A (R2), in: International Conference on Harmonization, IFPMA, Geneva, 2003;(b) K.K. Hotha, S. Phani, K. Reddy, V.K. Raju, L.K. Ravindranath, Forced degradation studies: practical approach-overview of regulatory guidance and literature for the drug products and drug substances, Int. Res. J. Pharm. 4 (2013) 78-85. |
| [34] | M. Sankaran, C. Kumarasamy, U. Chokkalingam, P.S. Mohan, Synthesis, antioxidant and toxicological study of novel pyrimido quinoline derivatives from 4-hydroxy-3-acyl quinolin-2-one, Bioorg. Med. Chem. Lett. 20 (2010) 7147-7151. |
| [35] | H.Y. Zhang, Structure-activity relationships and rational design strategies for radical-scavenging antioxidants, Curr. Comput. Aided Drug Des. 1 (2005) 257-273. |
| [36] | R.W. Blakesley, Methods for preventing inhibition of nucleic acid synthesis by pyrophosphate, US6291164, 2001. |
| [37] | P. Ertl, B. Rohde, P. Selzer, Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties, J. Med. Chem. 43 (2000) 3714-3717. |