 2016, Vol.27
2016, Vol.27 



b School of Petrochemical Engineering, Changzhou University, Changzhou 213164, China;
c Department of Microbiology, China Pharmaceutical University, Nanjing 210009, China
The emergence and spread of multi-drug resistant bacteria have become a serious threat to public health [1]. In particular,multidrug resistant Gram-positive bacteria including methicillin-resistant Staphylococcus aureus (MRSA) [2],penicillin-resistant Streptococcus pneumoniae (PRSP) [3],and vancomycin-resistant Enterococci (VRE) [4] represent major concerns. There is,therefore,an urgent need to identify and develop novel antibiotics with modes of action that are distinct from those of established classes.
Pleuromutilin (1,Fig. 1),a diterpenoid natural product with a fused 5-6-8 tricyclic skeleton,was first isolated in 1951 from two basidiomycete species [5]. It displays modest in vitro antibacterial activity against Gram-positive pathogens and mycoplasmas. Further studies have shown that the pleuromutilin class of antibiotics selectively inhibits bacterial protein synthesis by specifically targeting the 50s subunit of the bacterial ribosome and displays no cross-resistance with antibiotics currently in clinical use [6]. The distinct mode of action of pleuromutilin has made it an attractive target in the development of novel antibiotics for the treatment of multi-drug resistant bacterial infections.
Since the 1960s,numerous semisynthetic pleuromutilin analogs have been prepared and evaluated,and it was recognized that pleuromutilin derivatives containing a sulfide linkage in the C-14 side chain demonstrated superior antibacterial activity [7]. Of the derivatives generated,tiamulin [8] (2,Fig. 1) and valnemulin [9] (3,Fig. 1) were successfully developed as veterinary medicine. In 2007,retapamulin [10] (4,Fig. 1),a novel pleuromutilin derivative,was first approved forhuman use as a topical antimicrobial agent to treat skin infections. Other pleuromutilin derivatives in clinical trials include Nabriva Therapeutics’ BC-7013 [11] (5,Fig. 1) and BC-3781 [12] (6,Fig. 1). Especially,BC-3781 has completed phase II clinical trial for the systemic treatment of acute bacterial skin and skin structure infections (ABSSSI) and acquired Qualified Infectious Disease Product (QIDP) as well as fast track status designation.

|
Download:
|
| Fig. 1.uctures of pleuromutilin and its derivatives. | |
4H-Pyran-4-one and pyridin-4-one ring systems are very attractive moieties in medicinal chemistry because of their occurrence in a variety of bioactive compounds [13]. Existing studies have proved their wide range of biological activities such as antiproliferative,antibacterial,and antimalarial activity [14,15,16]. Based on these interesting biological activity and previous SAR of pleuromutilins,a series of novel thioether pleuromutilin derivatives bearing 4H-pyran-4-one and pyridin-4-one moieties in the C-14 side chain were designed,synthesized,and evaluated for their antibacterial activity. Herein,we described the details of this study.
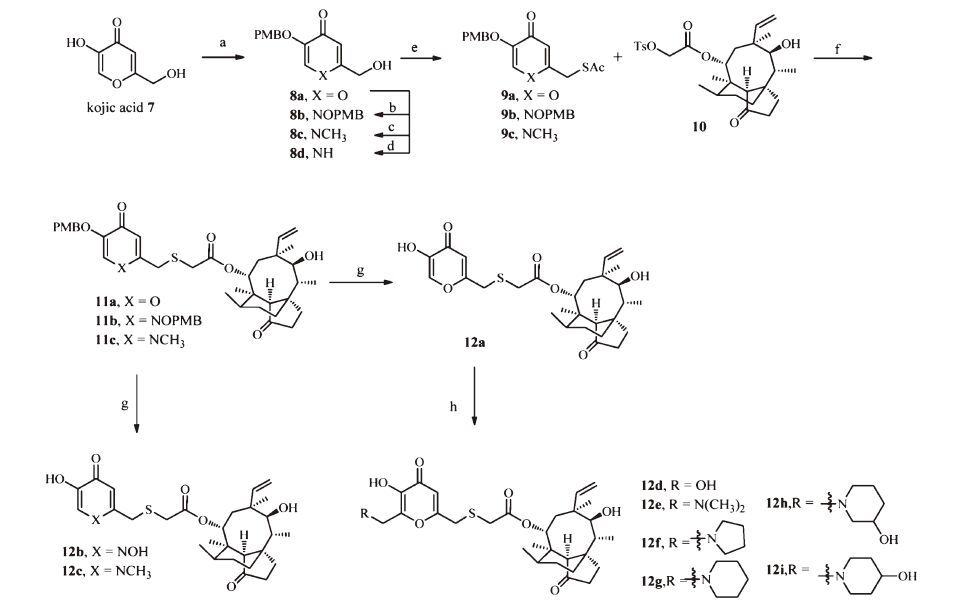
2. ExperimentalThe synthesis of compounds 12a-12i is shown in Scheme 1. Commercially available kojic acid was protected with 4- methoxybenzyl group at C-5 position to give compound 8a,which was transformed to 8b-8d by reported method [17]. Mesylation of the 2-hydroxymethyl group in compounds 8a-8c followed by nucleophilic substitution with potassium thioacetate gave compounds 9a-9c in =%-62% yield over 2 steps. Hydrolysis of the thioacyl groups in compounds 9a-9c revealed the corresponding thiol anions that reacted with mutilin 14-tosyloxyacetate 10 under basic condition to give compounds 11a-11c in 57%-68% yield. Deprotection of the 4-methoxybenzyl group in compounds 11a- 11c by trifluoroacetic acid yielded compounds 12a-12c in =%- 70% yield. Treatment of compound 12a with formaldehyde solution furnished compound 12d. Mannich reaction of compound 12a with corresponding amines afforded compounds 12e-12i in 46%-59% yield.
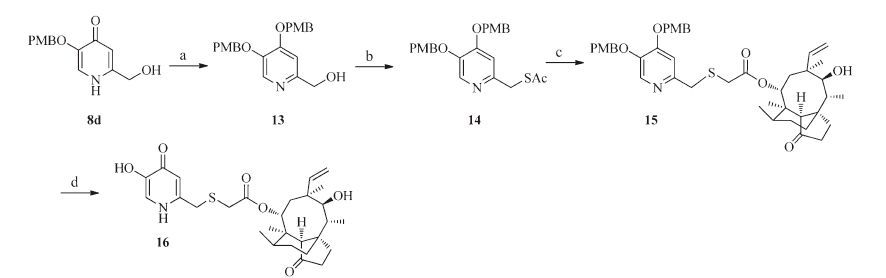
As shown in Scheme 2,compound 16 was prepared from compound 8d. Reaction of compound 8d with PMBCl and K2CO3 in DMF at 70 ℃ afforded compound 13 in 68% yield and N-4- methoxybenzylated byproduct was isolated in 12% yield. Compound 13 was then converted to compound 16 by the same procedures as described for compounds 12a-12c.

|
Download:
|
| Scheme 1.Synthesis of compounds 12a–12i. Reagents and conditions: (a) PBMCl, K2CO3, DMF, 65 ℃, 8 h; (b) (i) NH2OH-HCl, CH3COONa, N-methyl pyrrolidone, 70 ℃, 12 h; (ii) PBMCl, K2CO3, DMF, 65 ℃, 6 h; (c) CH3NH2/alcohol solution, 70 ℃, 6 h; (d) 25% aqueous NH4OH, 60 ℃, 12 h; (e) (i) MsCl, TEA, CH2Cl2, 0 ℃–r.t., 12 h; (ii) KSAc, DMF, 40 ℃, 2–4 h; (f) 5% aqueous NaOH, MeOH/CH2Cl2, r.t.; (g) CF3COOH, CH2Cl2, 0 ℃–r.t., 4–6 h; (h) 37% aqueous CH2O, NaOH, MeOH, r.t. or 37% aqueous CH2O, R1R2NH, MeOH, r.t. | |

|
Download:
|
| Scheme 2.Synthesis of compound 16. Reagents and conditions: (a) PBMCl, K2CO3, DMF, 65 ℃, 8 h; (b) (i) MsCl, TEA, CH2Cl2, 0 ℃–r.t., 12 h; (ii) KSAc, DMF, 40 ℃, 4 h; (c) 5% aqueous NaOH, mutilin 14-tosyloxyacetate 10, MeOH/CH2Cl2, r.t.; (d) CF3COOH, CH2Cl2, 0 ℃–r.t., 6 h. | |
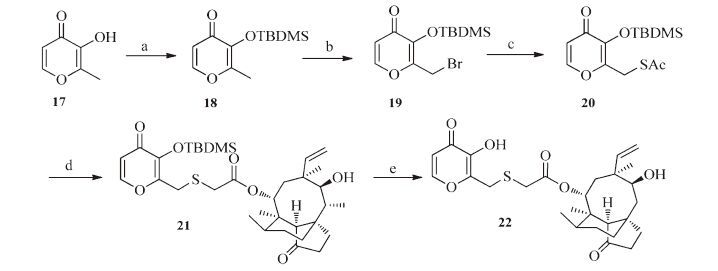
As shown in Scheme 3,the synthesis of compound 22 started from maltol (compound 17),which was converted to compound 19 by TBDMS-protection and subsequent bromination. Nucleophilic substitution of compound 19 with potassium thioacetate followed by hydrolysis of the thioacyl group and reaction with 10 gave compound 21. Deprotection of the TBDMS group in compound 21 afforded compound 22 in quantitative yield.

|
Download:
|
| Scheme 3.Synthesis of compound 22. Reagents and conditions: (a) TBDMSCl, imidazole, CH2Cl2, 0 ℃–r.t., 6 h; (b) NBS, AIBN, CCl4, 60 ℃, N2, 8 h; (c) KSAc, DMF, 40 ℃, 5 h; (d) mutilin 14-tosyloxyacetate 10, 5% aqueous NaOH, MeOH/CH2Cl2, r.t.; (e) TBAF/THF, 0 ℃–r.t., 5 h. | |
The synthesis of compound 28 is illustrated in Scheme 4. The primary alcohol of kojic acid (compound 7) was protected with the tetrahydropyranyl group to give compound 23,which was treated with formaldehyde solution to yield compound 24. Protection of the C-5 hydroxyl group afforded compound 25 which was converted to compound 28 following procedures similar to those described for compounds 12a-12c.

|
Download:
|
| Scheme 4.Synthesis of compound 28. Reagents and conditions: (a) 3,4-dihydro-2H-pyran, 0.1% PTSA-H2O, CH2Cl2, r.t., 4 h; (b) 37% aqueous CH2O, NaOH, MeOH, r.t.; (c) PBMCl, K2CO3, DMF, 65 ℃, 8 h; (d) (i) MsCl, TEA, CH2Cl2, 0 ℃–r.t., 12 h; (ii) KSAc, DMF, 40 ℃, 4 h; (e) 5% aqueous NaOH, mutilin-14-tosyloxyacetate 10, MeOH/CH2Cl2, r.t.; (f) CF3COOH, CH2Cl2, 0 ℃–r.t., 6 h. | |
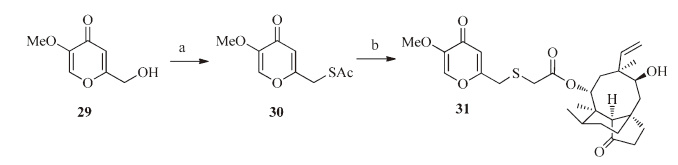
As shown in Scheme 5,compound 31 was synthesized from compound 29 by the same procedures as described for compounds 12a-12c.

|
Download:
|
| Scheme 5.Synthesis of compound 31. Reagents and conditions: (a) (i) MsCl, TEA, CH2Cl2, 0 ℃–r.t., 12 h; (ii) KSAc, DMF, 35 ℃, 4 h; (b) 5% aqueous NaOH, mutilin 14- tosyloxyacetate 10, MeOH/CH2Cl2, r.t. | |
The in vitro antibacterial activities of the target compounds were tested against clinically isolated Gram-positive strains with retapamulin and BC-3781 as positive controls. Theminimal inhibitory concentration (MIC) values were determined using agar dilution method according to CLSI guidelines [18]. The results were summarized in Table 1. It was interesting that compound 12a exhibited very potent antibacterial activity (MIC = 0.031-0.25 mg/mL),whereas compound 12b,bearing N-hydroxylpyridin-4-one moiety,almost lost activity. Replacement of the N-hydroxyl group in compound 12b with a methyl group or hydrogen atom restored some of the activity (compounds 12c and 16 vs. compound 12b). This suggested that 4H-pyran-4-one moiety is very important for maintaining antibacterial activity. Further modifications were then focused on compound 12a. Introduction of hydroxymethyl or amino groups onto the pyranone ring of compound 12a gave compounds 12d-12i,which displayed moderate to good activity. Compound 22,the regioisomer of compound 12a,showed a 2-4-fold decreased antibacterial activity,whereas compound 28 exhibited comparable activity to its regioisomer,compound 12d. O-Methylation of the hydroxyl group in the 4H-pyran-4-one moiety of compound 12a gave compound 31 with 2-4-fold reduced activity,indicating the hydroxyl group was essential for potent antibacterial activity. Among all the derivatives,compounds 12a,12d,and 28 aremost potent in this series,exhibiting antibacterial activity comparable to or slightly better than retapamulin and BC-3781.
|
|
Table 1 Minimum inhibitory concentration of pleuromutilin derivatives. |

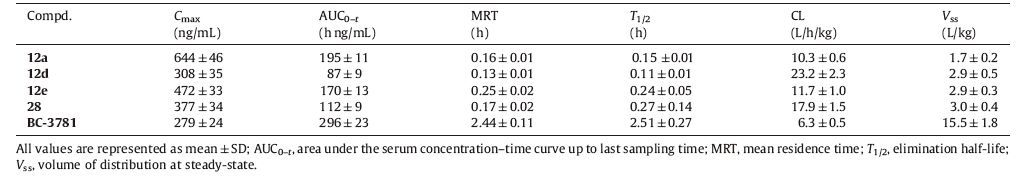
Compounds 12a,12d,12e and 28 were then selected for pharmacokinetic study. Cassette-dosing experiments were performed on these compounds with clinical investigational drug BC- 3781 included for comparison. Results of this study are summarized in Table 2.
|
|
Table 2 Pharmacokinetic parameters of compounds 12a, 12d, 12e, 28 and BC-3781 by cassette dosing at 2 mg/kg in rats (i.v., n = 3). |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
As shown in Table 2,while all the target compounds displayed a favorable maximum plasma concentration (Cmax),their plasma exposure levels (AUC0-t) were much lower than that of BC-3781. An elimination half-life (T1/2) and a mean residence time (MRT) of below 15 min suggested that these compounds underwent a rapid clearance from plasma after intravenous administration. As the PK profiles were not favorable,further investigation and structural optimization are still ongoing.
4. ConclusionBy incorporation of 4H-pyran-4-one and pyridin-4-one moieties into the C-14 side chain,a series of novel pleuromutilin derivatives were designed and synthesized. Structure-activity relationship studies showed that 4H-pyran-4-one moiety is favored over pyridin-4-one. Among the series,compounds 12a,12d,and 28 exhibited excellent antibacterial activity against clinically isolated Gram-positive strains including MRSA,MRSE,and PRSP. Cassette-dosing experiments showed that theses novel pleuromutilin derivatives underwent a rapid clearance after intravenous administration. Further investigation and structural modifications are needed to improve their PK profiles.
5. Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.09.019.
| [1] | B. Spellberg, R. Guidos, D. Gilbert, et al., The epidemic of antibiotic-resistant infections: a call to action for the medical community from the infectious diseases society of America, Clin. Infect. Dis. 46 (2008) 155-164. |
| [2] | H.W. Boucher, G.H. Talbot, J.S. Bradley, et al., Bad bugs, no drugs: no ESKAPE! An update from the infectious diseases society of America, Clin. Infect. Dis. 48 (2009) 1-12. |
| [3] | T. Alexander, Antibiotic-resistance in Streptococcus pneumoniae, Clin. Infect. Dis. 24 (1997) S85-S88. |
| [4] | Y. Cetinkaya, P. Falk, C.G. Mayhall, Vancomycin-resistant Enterococci, Clin. Microbiol. Rev. 13 (2000) 686-707. |
| [5] | F. Kavanagh, A. Hervey, W.J. Robbins, Antibiotic substances from basidiomycetes. VⅢ. Pleurotus multilus (Fr.) sacc. and Pleurotus passeckerianus pilat, Proc. Natl. Acad. Sci. U. S. A. 37 (1951) 570-574. |
| [6] | L.A. Hodgin, G. Högenauer, The mode of action of pleuromutilin derivatives, Eur. J. Biochem. 47 (1970) 527-533. |
| [7] | H. Egger, H. Reinshagen, New pleuromutilin derivatives with enhanced antimicrobial activity. Ⅱ. Structure-activity correlations, J. Antibiot. 29 (1976) 923-927. |
| [8] | G. Laber, E. Schütze, In vivo efficacy of 81.723 hfu, a new pleuromutilin derivative against experimentally induced airsacculitis in chicks and turkey poults, Antimicrob. Agents Chemother. 7 (1975) 517-521. |
| [9] | P.C.T. Hannan, H.M. Windsor, P.H. Ripley, In vitro susceptibilities of recent field isolates of Mycoplasma hyopneumoniae and Mycoplasma hyosynoviae to valnemulin (Econor®), tiamulin and enrofloxacin and the in vitro development of resistance to certain antimicrobial agents in Mycoplasma hyopneumoniae, Res. Vet. Sci. 63 (1997) 157-160. |
| [10] | R.S. Daum, S. Kar, P. Kirkpatrick, Retapamulin, Nat. Rev. Drug Discov. 6 (2007) 865-866. |
| [11] | R. Novak, Are pleuromutilin antibiotics finally fit for human use? Ann. N. Y. Acad. Sci. 1241 (2011) 71-81. |
| [12] | W.T. Prince, Z. Ivezic-Schoenfeld, C. Lell, et al., Phase Ⅱ clinical study of BC-3781, a pleuromutilin antibiotic, in treatment of patients with acute bacterial skin and skin structure infections, Antimicrob. Agents Chemother. 57 (2013) 2087-2094. |
| [13] | Y.S. Lee, J.H. Park, M.H. Kim, S.H. Seo, H.J. Kim, Synthesis of tyrosinase inhibitory kojic acid derivative, Arch. Pharm. 339 (2006) 111-114. |
| [14] | B.V.S. Reddy, M.R. Reddy, C.H. Madan, K.P. Kumar, M.S. Rao, Indium(Ⅲ) chloride catalyzed three-component coupling reaction: a novel synthesis of 2-substituted aryl(indolyl)kojic acid derivatives as potent antifungal and antibacterial agents, Bioorg. Med. Chem. Lett. 20 (2010) 7507-7511. |
| [15] | J.M. Bueno, P. Manzano, M.C. García, et al., Potent antimalarial 4-pyridones with improved physico-chemical properties, Bioorg. Med. Chem. Lett. 21 (2011) 5214-5218. |
| [16] | Y.H. Chen, P.J. Lu, C. Hulme, A.Y. Shaw, Synthesis of kojic acid-derived copperchelating apoptosis inducing agents, Med. Chem. Res. 22 (2013) 995-1003. |
| [17] | M.F. Brown, M.J. Mitton-Fry, J.T. Arcari, et al., Pyridone-conjugated monobactam antibiotics with Gram-negative activity, J. Med. Chem. 56 (2013) 5541-5552. |
| [18] | CLSI, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard M07-A9, 9th ed., Clinical and Laboratory Standards Institute, Wayne, 2012. |