 2016, Vol.27
2016, Vol.27 



b Division of Anti-tumor Pharmacology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;
c Department of Medicinal Chemistry, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;
d Pharmacology and Safety Evaluation Key Laboratory of Tibetan Medicine in Qinghai Province, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining 810008, China
Angiogenesis,the formation of new blood vessels from preexisting vessels,is a normal and indispensable process in growth and development as well as in wound healing and female reproductive cycling [1, 2]. Pathological angiogenesis has been correlated with a variety of diseases,such as retinopathies,diabetes,rheumatoid arthritis,psoriasis,and cancers [3, 4]. Tumor angiogenesis is vital for tumor growth as it is a fundamental step in the growth and metastasis of cancer [5],making angiogenesis inhibition a promising therapeutic strategy against cancer.
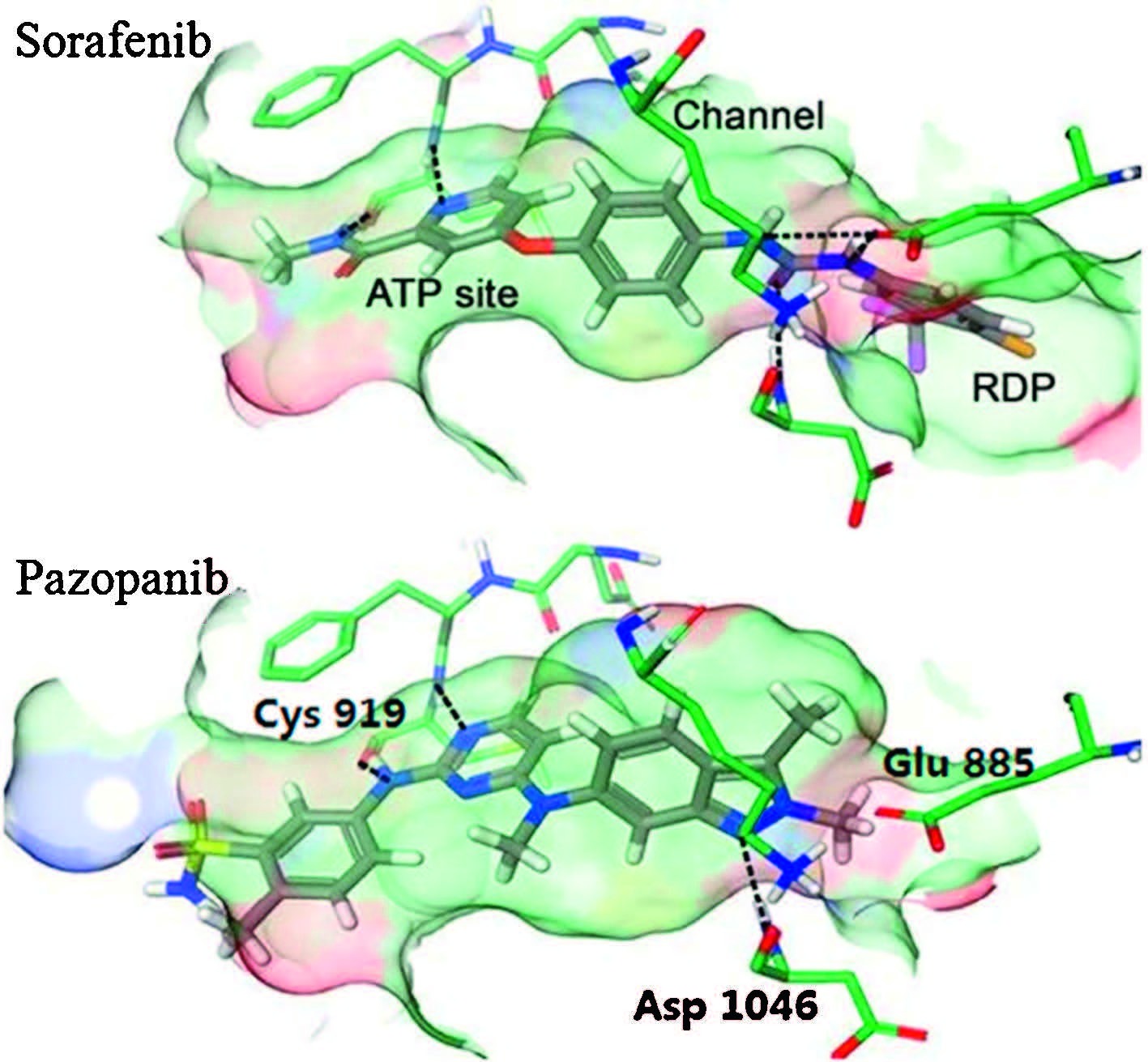
VEGFs and their receptors (especially VEGFR-2) are very important in the direct regulation of angiogenesis,and the overexpression of VEGFR-2 has been closely implicated in the angiogenesis of solid tumors [6]. Neutralization of the circulating VEGF by antibodies and inhibition of VEGF receptor tyrosine kinases with small molecules have been found to effectively inhibit angiogenesis,tumor progression and dissemination [7]. Enormous efforts have been made on the development of drugs targeting VEGFR-2,leading to several launched anticancer drugs,such as sunitinib [8, 9],sorafenib [10],vandetanib [11],pazopanib [12] and axitinib [13]. Nevertheless,intolerable adverse effects that cause dose reductions and treatment discontinuations may potentially negate the life-prolonging effects of these drugs [14],indicating an unmet need for safer VEGFR-2 inhibitors with high efficacy. As a part of our continuing program on VEGFR-2 inhibitors,we reported here the discovery and structural optimization of biphenylurea derivatives as new VEGFR-2 inhibitors. Sorafenib and pazopanib are now being used in clinic for the treatment of advanced renal cell carcinoma (RCC). A close look of their binding modes may provide some useful information; in general,both molecules bind to the ATP binding site,penetrated into the extended hydrophobic pocket and filled the RDP (regulatory domain pocket) region,locking the protein in a "DFG-out" conformation (Fig. 1); residing in the ATP binding site,Cys919 forms H-bonds with the pyridine nitrogen and amide N-H of sorafenib and the anilinopyrimidine scaffold of pazopanib. Glu885 is served as a H-bond donor to both molecules; Asp1046,however,only interacted with sorafenib [15].

|
Download:
|
| Fig. 1.Binding models of sorafenib (PDB ID: 4ASD) and pazopanib with VEGFR-2. Reproduced with the permission from Ref. [15]. | |
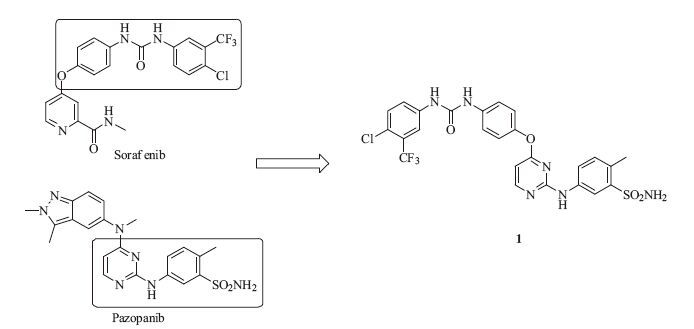
Direct H-bonds with Glu885 and Asp1046 were considered a significant opportunity for better selectivity,and greater molecular size in the RDP site would cause a reduction of efficacy [15]. We envisioned that an additional hydrogen bond to the Asp1046 by grafting the biphenylurea moiety from sorafenib to the anilinopyrimidine structure of pazopanib would hopefully provide a new scaffold with high selectivity. Tuning the size of the 3-chloro-4- (trifluoromethyl)benzene ring may yield better efficacy (Fig. 2). To our delight,compound 1 inhibited VEGFR-2 at the enzymatic level with an IC50 value of 69 nmol/L. Inspired by this promising result,we then synthesized a series of biphenylurea analogs of 1 to investigate the structure-activity relationships (SARs).

|
Download:
|
| Fig. 2.Design of hybrid compound 1. | |
The synthesis of analogs was illustrated in Scheme 1. 2,3 (or 4,6)-Dichloropyrimidine (2 and 3) were coupled with substituted p-nitrophenol to give intermediates 4-7 which were subsequently reduced to corresponding amine 8-11. Ureas 12-19 were prepared through a well-established method [16]. All the derivatives (1,20- 40) were prepared through an acid-catalyzed SNAr reaction of urea intermediates with substituted anilines in the final step. All derivatives listed in Tables 1-3 were evaluated for their enzymatic activities against VEGFR-2 and cellular activities against adenocarcinomic human alveolar basal epithelial cells (A549) and human microvascular endothelial cell (HMEC-1).

|
Download:
|
| Scheme 1.Synthesis of compounds 1, and 20–40. Reagents and conditions: (a) substituted p-nitrophenol, aq. sodium hydroxide, acetone, 0–80 ℃; (b) Fe, aq. ammonium chloride, ethanol, 70 ℃; (c) substituted anilines, triphosgene, triethyl amine, methylene chloride, r.t.; and (d) substituted anilines, 36% HCl, isopropanol, sealed tube, 100 ℃. | |
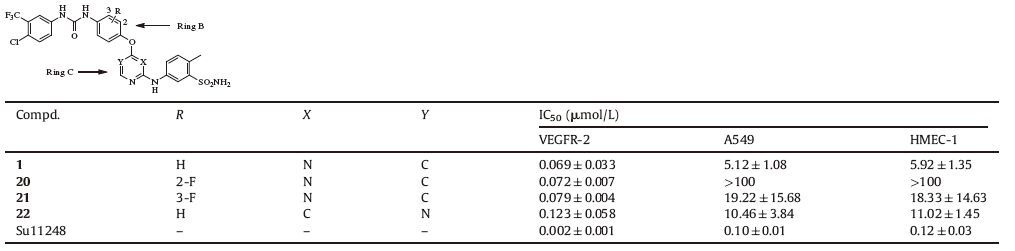
We first explored the effect of variation in rings B and C of compound 1 (Table 1). Incorporation of F atom on an aromatic ring could increase lipophilicity,mask a potential metabolic site and offer a polarized C-F bond that may participate in binding interaction [17]. Regorafenib,for example,was invented as a fluoro derivative of sorafenib with better activities. The derivatives with fluoro substitution on the B ring (20 and 21) were synthesized. Unfortunately,no improvement of activity was observed. We also altered the substitution pattern of the pyrimidine ring to move the nitrogen closer to the amino acid residues in the hinge region,hoping for an extra interaction. Compound 22,the 4,6-disubstitued pyrimidine analog of 1,was synthesized and examined. The result indicated that 22 displayed a 1.8-fold reduction in enzymatic activity (IC50: 123 nmol/L). These facts implied that unmodified rings B and C in compound 1 were favorable for maintaining activity.
|
|
Table 1 SAR of ring B and ring C. |
|
|
Table 2 SAR of 2-substituents on the pyrimidine ring. |

Next,we investigated the effect of substitution pattern of the aniline at pyrimidine ring on the activity. At first,a set of five sulfonamides including four meta-sulfonamides (23,25-27) and one para-sulfonamide (24) were explored. Although these derivatives exhibited high inhibitory potency against VEGFR-2 with IC50 values ranging from 4 nmol/L to 69 nmol/L,they were inactive on cancer cell proliferation assay. Then,the synthesis of a series of derivatives with various substituted anilines on pyrimidine ring led to 34 (3,4-dimethyl substitution) which exhibited potent activities in both enzymatic and cellular assays (IC50 values of 7 nmol/L,2.88 μmol/L and 2.81 μmol/L for VEGFR-2,A549 and HMEC-1,respectively). Interestingly,unsubstituted aniline derivative 36 showed high potency against VEGFR-2 and A549 (IC50: 5 nmol/L and 8.4 μmol/L,respectively) but low potency against HMEC-1 (IC50: 46.28 μmol/L). Taken together,the aniline group at the 2-position of pyrimidine was tolerable to modification. 3,4- dimethoxy substituted analog was proven to be the best in this series of compounds at both enzymatic and cellular levels,and therefore further investigation was focused on the 3,4-dimethoxy substituted analogs.
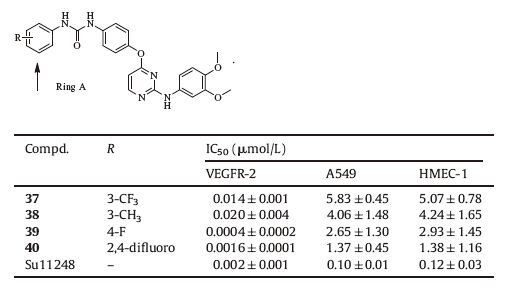
Finally,a set of analogs of 34 was prepared to explore the influence of the substituent on ring A,and this exploration led to analogs 37-40 as shown in Table 3. Among them,39 exhibited high enzymatic inhibitory activity with an IC50 value of 0.4 nmol/L,which was one of the most potent VEGFR-2 inhibitors as far as we knew. The privileged compound 39 was further examined in HUVEC proliferation assay and it exhibited high inhibitory potency with an IC50 value of 20.36 nmol/L.
|
|
Table 3 SAR of ring A. |
Compound 39 was then profiled against a panel of tyrosine kinases. The result demonstrated that 39 inhibited a distinctive,narrow range of kinases at pharmacologically relevant concentrations (as shown in Table 4). In addition to its high inhibitory potency on VEGFR-2,39 also exhibited great potency against VEGFR-1,PDGFR-α and PDGFR-β with IC50 values of 3.4,4.4 and 3.6 nmol/L,respectively. Simultaneous inhibition of these distinct proangiogenic receptors may enhance the antitumor efficacy of 39 and overcome resistance to VEGF- and VEGFR-2-pathway targeted agents. Moreover,39 showed high selectivity over RET (1574-fold,IC50: 629.7 nmol/L) and excellent selectivity (>25,000-fold) over ErbB2,ErbB4,EGFR,EGFR (T790M/L858R),ABL,EPH-A2,FGFR-1,FGFR-2 and IGF-1R.
|
|
Table 4 Kinase-selectivity profiling of compound 39. |

{kind=link}
{kind=link}
{kind=link}
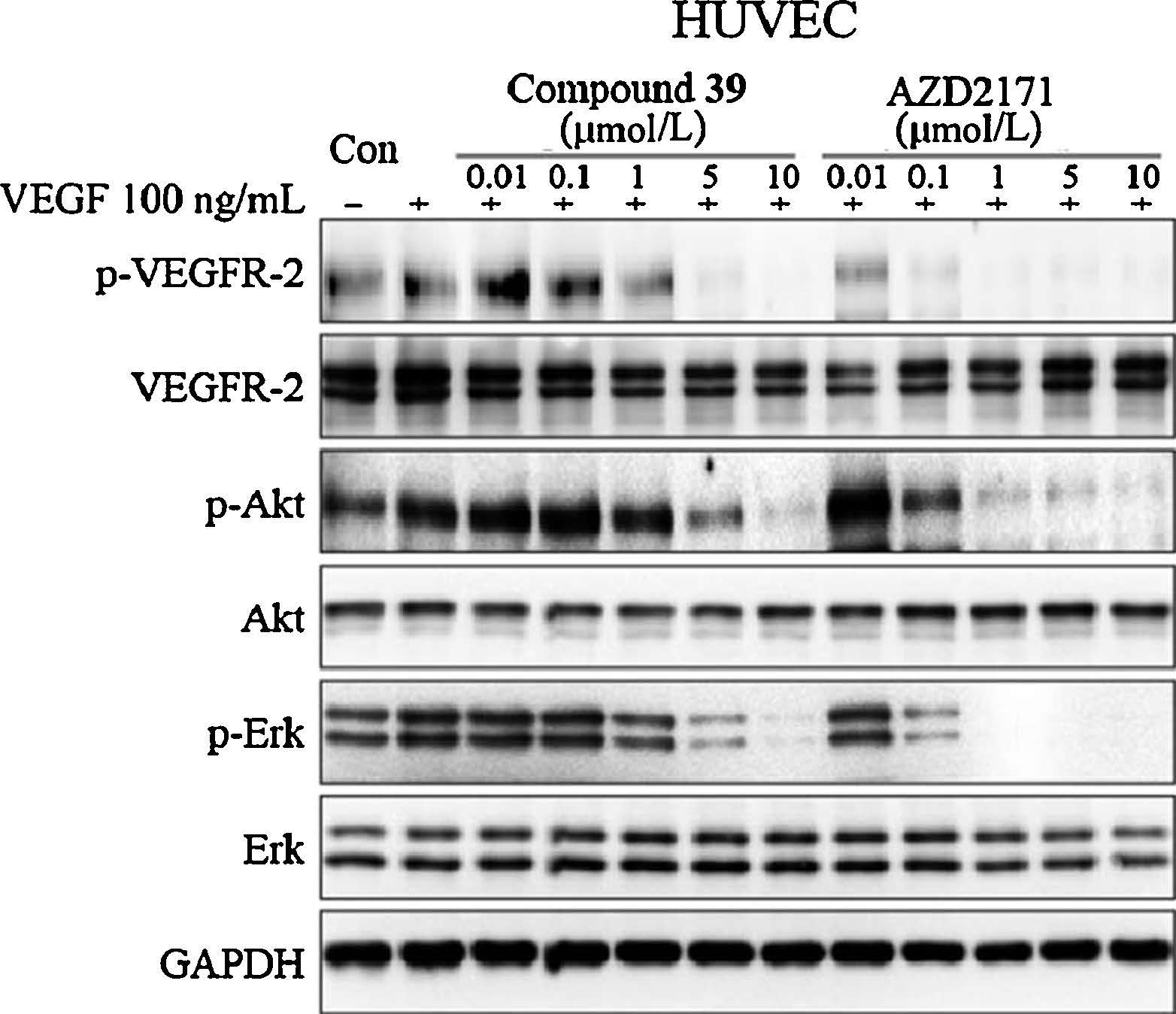
Subsequently,inhibition of VEGF stimulated of cell proliferation of HUVEC by 39 (positive control: AZD2171 [18]) was tested. As shown in Fig. 3,the VEGF-induced phosphorylation of VEGFR2 (p-VEGFR-2) was substantially decreased in the presence of 39 in a dose-dependent manner. Meanwhile,Akt and Erk,the downstream effector of VEGF/VEGFR,were also attenuated in a dose-dependent manner.

|
Download:
|
| Fig. 3.Inhibition of VEGFR-2 phosphorylation and related signaling pathways by compound 39. HUVEC cells were starved for 12 h and treated with indicated concentration of compounds for 2 h and then stimulated by 100 ng/mL VEGF for 15 min. Cells were harvested and subjected to Western blot analysis. | |
{kind=link}
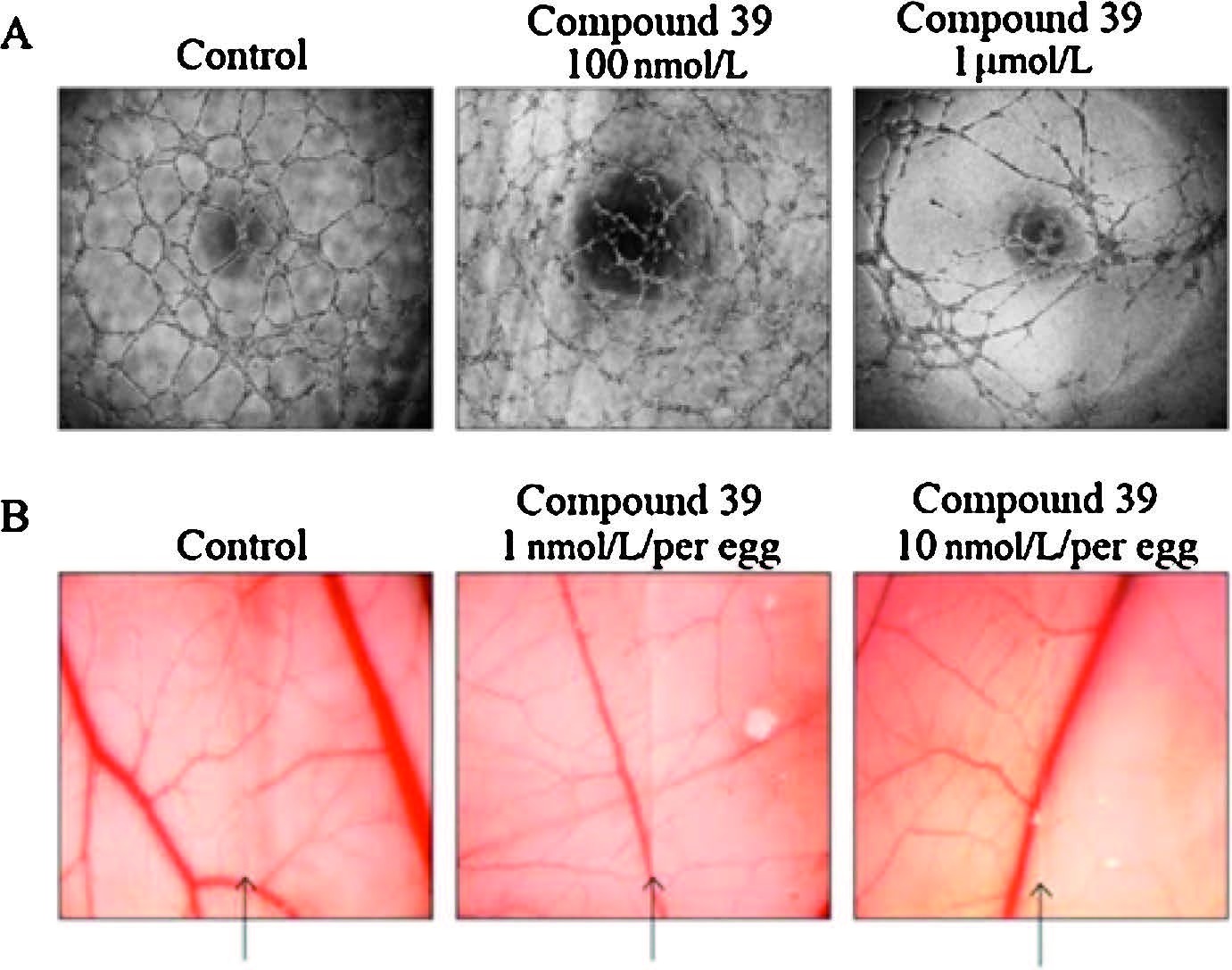
Eventually,the anti-angiogenic effect of 39 was also examined. In vitro tube formation assay of HUVECs showed that HUVECs formed a complete network structure within 6 h. After treatment of 39,the tube formation was suppressed in a dosedependent manner (Fig. 4A). Further examination of the antiangiogenic effect of 39 was performed,using chick chorioallantoic membrane (CAM) assay,a method to evaluate in vivo neovascularization. The results (Fig. 4B) demonstrated that vascularization in chick embryos was significantly inhibited when the embryos were treated with 39 (1 and 10 nmol per egg). These results proved that 39 possessed potent antiangiogenic activities both in vitro and in vivo.

|
Download:
|
| Fig. 4.Antiangiogenesis activities of compound 39. (A) Compound 39 inhibited in vitro tube formation of HUVECs. HUVECs containing with or without indicated concentration of compound were seeded in 96-well plates which were coated with Matrigel. After 6 h, the tube was examined with an inverted phase contrast microscope. (B) Compound 39 showed anti-angiogenesis in CAM model. Glasscover-slip saturated with or without compound 39 was placed on the right side of per field. Arrows indicated the edge line of the coverglass. | |
{kind=link}
In summary,we designed and synthesized a series of compounds as VEGFR2 inhibitors. A highly potent VEGFR-2 inhibitor 39 was identified with an IC50 value of 0.4 nmol/L. Kinase profiling study indicated that 39 also inhibited three distinct proangiogenic receptors,VEGFR-1,PDGFR-α and PDGFR-β with high selectivity over other kinases. More importantly,39 effectively inhibited angiogenesis in both in vitro HUVEC tube formation assay and in vivo CAM assay. Therefore,a new VEGFR-2 inhibitor scaffold and an understanding of their SARs offer promising opportunities to develop more effective anticancer agents.
5. AcknowledgmentsWe thank the National Natural Science Foundation of China (Nos. 81273365,81173080,81321092),National Science & Technology Major Project ‘‘Key New Drug Creation and Manufacturing Program’’ (Nos. 2012ZX09103101-024,2014ZX09304002-008-001),Chinese National Programs for High Technology Research and Development (No. 2012AA020302) and the Shanghai Science and Technology Commission (No. 12DZ1930802) for their financial support. The authors gratefully thank the Proceedings of the National Academy of Sciences USA for their generous permission to reprint Fig. 1.
| [1] | P. Carmeliet, Angiogenesis in health and disease, Nat. Med. 9 (2003) 653-660. |
| [2] | N. Ferrara, R.S. Kerbel, Angiogenesis as a therapeutic target, Nature 438 (2005) 967-974. |
| [3] | Z.K. Otrock, J.A. Makarem, A.I. Shamseddine, Vascular endothelial growth factor family of ligands and receptors: review, Blood Cells. Mol. Dis. 38 (2007) 258-268. |
| [4] | A. Garofalo, A. Farce, S. Ravez, et al., Synthesis and structure-activity relationships of (Aryloxy) quinazoline ureas as novel, potent, and selective vascular endothelial growth factor receptor-2 inhibitors, J. Med. Chem. 55 (2012) 1189-1204. |
| [5] | K. Sanphanya, S.K. Wattanapitayakul, S. Phowichit, V.V. Fokin, O. Vajragupta, Novel VEGFR-2 kinase inhibitors identified by the back-to-front approach, Bioorg. Med. Chem. Lett. 23 (2013) 2962-2967. |
| [6] | H.M.W. Verheul, H.M. Pinedo, Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition, Nat. Rev. Cancer 7 (2007) 475-485. |
| [7] | R.A. Brekken, J.P. Overholser, V.A. Stastny, et al., Selective inhibition of vascular endothelial growth factor (VEGF) receptor 2 (KDR/Flk-1) activity by a monoclonal anti-VEGF antibody blocks tumor growth in mice, Cancer Res. 60 (2000) 5117-5124. |
| [8] | X.W. Zhao, D. Liu, S.L. Luan, et al., Synthesis and biological evaluation of substituted 1,2,3-benzotriazines and pyrido[3,2-d]-1,2,3-triazines as inhibitors of vascular endothelial growth factor receptor-2, Bioorg. Med. Chem. 21 (2013) 7807-7815. |
| [9] | T.A. Fong, L.K. Shawver, L. Sun, et al., SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types, Cancer Res. 59 (1999) 99-106. |
| [10] | S. Wilhelm, C. Carter, M. Lynch, et al., Discovery and development of sorafenib: a multikinase inhibitor for treating cancer, Nat. Rev. Drug Discov. 5 (2006) 835-844. |
| [11] | H. Commander, G. Whiteside, C. Perry, Vandetanib: first global approval, Drugs 71 (2011) 1355-1365. |
| [12] | P.A. Harris, A. Boloor, M. Cheung, et al., Discovery of 5-[[4-[(2,3-dimethyl-2Hindazol-6-yl)methylamino]-2-pyrimidinyl] amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor, J. Med. Chem. 51 (2008) 4632-4640. |
| [13] | T.H. Ho, E. Jonasch, Axitinib in the treatment of metastatic renal cell carcinoma, Future Oncol. 7 (2011) 1247-1253. |
| [14] | B. Blanchet, B. Billemont, S. Barete, et al., Toxicity of sorafenib: clinical and molecular aspects, Expert Opin. Drug Saf. 9 (2010) 275-287. |
| [15] | M. McTiguea, B.W. Murray, J.H. Chen, et al., Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors, Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 18281-18289. |
| [16] | T.B. Norsten, K. Chichak, N.R. Branda, Strong and directed association of porphyrins and iron(terpyridine)s using hydrogen bonding and ion pairing, Tetrahedron 58 (2002) 639-651. |
| [17] | A.V. Razgulin, S. Mecozzi, Binding properties of aromatic carbon-bound fluorine, J. Med. Chem. 49 (2006) 7902-7906. |
| [18] | S.R. Wedge, J. Kendrew, L.F. Hennequin, et al., AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer, Cancer Res. 65 (2005) 4389-4400. |