 2016, Vol.27
2016, Vol.27 



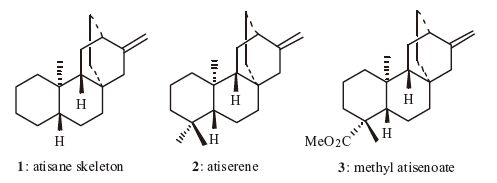
A large number of dierpenes with the atisane skeleton 1 have been isolated from different natural sources [1, 2],such as atiserene 2 and methyl atiseneoate 3 (Fig. 1). Many of these tetracyclic diterpenoids display a wide range of interesting biological activities including antimicrobial [3],antitumor [4] and antifeedant [5]. Due to the unique structure and significant biological activities,atisane diterpenes have attracted attention from the synthetic community for decades. A large part of this work has been based on a homoally-homoally radical rearrangement reaction [6] or an intramolecular double Michael reaction [7, 8, 9] to elaborate the bicyclo[2.2.2]octane moiety of the atisane framework. By using these strategies,the synthesis of several atisane ditepenes,such as atiserene [10],methyl atiseneoate [11, 12, 13],methyl gummiferolate [14, 15] and serofendic acids A and B [16],have been completed.

|
Download:
|
| Fig. 1. Selected atisane diterpenes. | |
{kind=link}
We have recently described a concise and efficient synthetic approach to construct the bicyclo[2.2.2]octane ring system,characteristic of atisane diterpenes,which was based on an oxidative dearomatization/IMDA cycloaddition [17]. As a continuation of this work,we wish to report here a convergent tactics for preparation of the tetracyclic carbon skeleton of atisane diterpenes,which is a complement of the existing strategies for the synthesis of this type of natural products. The synthetic pathway is outlined in Schemes 1 and 2.
2. ExperimentalAll reactions were carried out under argon unless otherwise stated. All the chemicals were purchased from commercial sources and were used without further purification. THF,toluene were distilled from sodium-benzophenone; dichloromethane was distilled from calcium hydride; methanol was distilled from Mg/I2. Column chromatography was performed using 200-300 mesh silica get. HR MS spectra were obtained with a Bruker BioTOFQ mass spectrometer; NMR spectra were acquired on a Varian INOVA-400/54 or Agilent DD2-600/54 instrument,using TMS as the internal standard.
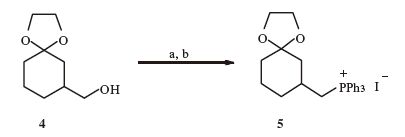
2.1. The preparation of compound 5To dry CH2Cl2 (20 mL) was added in this order: Triphenylphosphine (3.05 g,11.6 mmol),imidazole (0.763 g,11.6 mmol) and iodine (2.99 g,11.6 mmol). A solution of the known alcohol 4 (1 g,5.81 mmol) in 5 mL of dry CH2Cl2 was added and the mixture was stirred at room temperature for 3 h. Then the solvent was removed in vacuo and the residue was passed through a plug of silica with EtOAc/petroleum ether (1:100) to give the iodide as a yellow oil. This oil was dissolved in dry toluene (10 mL). To this solution was added triphenylphosphine (3.05 g,11.6 mmol),and the mixture was refluxed for 2 days. After being cooled down to room temperature and filtered through Celite,the solid was washed by petroleum ether (30 mL × 3) to give phosphonium salt 5 (1.64 g,52%) as a white solid. 5: 1H NMR (400 MHz,CDCl3): δ 7.91-7.86 (m,3H),7.82-7.78 (m,3H),7.73-7.68 (m,5H),7.33-7.30 (m,2H),3.97-3.57 (m,6H),2.12-2.04 (m,1H),1.70-1.66 (m,2H),1.57-1.49 (m,2H),1.46-1.31 (m,4H); 13C NMR (150 MHz,CDCl3): δ 134.87,134.85,133.32,133.25,130.21,118.32,117.75,107.65,63.86,41.95,33.66,32.86,30.60,28.24,27.92,22.36. HRMS (ESI-TOF): m/ z calcd. for (C27H30O2P+): 417.1978; found: 417.1989.
2.2. The preparation of compound 8 and 9To a stirred solution of the known ketal 7 (100 mg,0.420 mmol) in dry MeOH (1 mL) was added a solution of SmI2 (0.1 mol/l,4.2 mL,4.20 mmol) in dry THF at room temperature under argon. After the resulting mixture had been stirred for 30 min,the solvent was evaporated under reduce pressure. The residue was dissolved in water (5 mL) and concentrated HCl (1 mL),and then extracted with EtOAc (15 mL × 3). The combined organic extracts were washed successively with saturated aqueous NaHCO3 (20 mL),saturated aqueous Na2S2O3 (20 mL),and brine,dried over anhydrous MgSO4,filtered,and concentrated. The crude product was purified by column chromatography with EtOAc/petroleum ether (1:5-1:3) to give lactone 8 (18 mg,23%) as a yellow oil and alcohol 9 (62 mg,73%) as a yellow oil. 8: 1HMNR(400 MHz,CDCl3): d 6.60 (t,1H,J = 5.6 Hz),6.33 (d,1H,J = 5.6 Hz),4.57 (d,1H,J = 5.6 Hz),3.26 (t,1H,J = 2.0 Hz),2.75 (dd,1H,J = 4.8,6.8 Hz),2.31-2.29 (m,1H),2.28 (d,1H,J = 11.2 Hz),2.21 (d,1H,J = 11.2 Hz),1.89-1.86 (m,1H); 13C NMR (150 MHz,CDCl3): δ 208.60,175.40,135.70,134.48,72.79,48.39,46.32,43.60,42.56,25.63; 9: 1H MNR (400 MHz,CDCl3): δ 6.38 (t,1H,J = 5.2 Hz),6.10 (d,1H,J = 5.6 Hz),3.83 (s,3H),3.82 (s,1H),3.71 (s,1H),3.24 (t,1H,J = 2.0 Hz),2.97 (dd,1H,J = 3.6,6.4 Hz),2.25 (d,1H,J = 12.0 Hz),2.19-2.13 (m,2H),2.02-1.97 (m,2H); 13C NMR (150 MHz,CDCl3): δ 210.71,174.37,134.19,129.85,65.61,52.15,48.79,46.57,43.39,43.35,28.=. HRMS (ESI-TOF): m/z calcd. for (C10H10O3+Na+): 201.0528; found 201.0522.
2.3. The preparation of compound 10To an ice-cold solution of the alcohol 9 (50 mg,0.236 mmol) in dry CH2Cl2 (3 mL) was added 2,6-lutidine (41 mL,0.354 mmol) and TBSOTf (69 mL,0.307 mmol) sequentially,and the resulting reaction mixture was stirred at room temperature for 1 h. After being poured into 10 mL of saturated aqueous NaHCO3 at 0 ℃,the solution was extracted with CH2Cl2 (10 mL × 3) and the combined organic extracts were dried over anhydrous Na2SO4,filtered and concentrated in vacuo. The crude residue was purified by flash chromatography with EtOAc/petroleum ether (1:5-1:3) to give TBS ether as a colorless oil,which was dissolved in dry CH2Cl2 (2 mL). To this solution was added a solution of DIBALH (1 mol/L,1.0 mL,0.944 mmol) in dry toluene dropwise at 0 ℃ and the resulting mixture was still stirred at the same temperature for 2 h before it was quenched with saturated aqueous sodium potassium tartrate (5 mL). The solution was extracted with EtOAc (10 mL × 3) and the combined organic extracts were dried through anhydrous Na2SO4,filtered and evaporated in vacuo to give a residue,which was subjected to flash column chromatography via EtOAc/ petroleum ether (1:3-1:1) to deliver a colorless oil. To a solution of the oil in dry CH2Cl2 (3 mL) was added Dess-Martin periodinane (313 mg,0.708 mmol) and the mixture was stirred for 2 h at 0 ℃ prior to being quenched by the addition of saturated Na2S2O3 (5 mL). The mixture was extracted with CH2Cl2 (10 mL × 3). The organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified via flash chromatography with EtOAc/petroleum ether (1:5) to afford aldehyde 10 (35 mg,50%) as a colorless oil. 10: 1H NMR (400 MHz,CDCl3): δ 9.46 (d,1H,J = 2.8 Hz),6.38 (t,1H,J = 5.2 Hz),6.28 (d,1H,J = 5.6 Hz),3.87 (d,1H,J = 6.4 Hz),3.78 (d,1H,J = 6.8 Hz),3.26-3.25 (m,1H),2.85-2.82 (m,1H),2.11 (d,1H,J = 12.4 Hz),2.07 (d,1H,J = 7.2 Hz),1.95-1.92 (m,1H),0.90 (s,9H),0.09 (s,3H),0.08 (s,3H); 13C NMR (150 MHz,CDCl3): δ 210.17,201.51,135.21,129.99,65.37,52.03,48.40,46.88,42.66,25.71,25.15,18.15,-5.68,-5.73. HRMS (ESI-TOF): m/z calcd. for (C11H14O4+Na+): 233.0790; found 233.0790.
2.4. The preparation of compound 11To a stirred solution of the aldehyde 10 (100 mg,0.340 mmol) in DMF (5 mL) were sequentially added 2,20-bipyridine (79.6 mg,0.510 mmol),DABCO (112 mg,0.510 mmol),cupric acetate (102 mg,0.510 mmol),and the resulting mixture was heated at 70 ℃ for 6 h. After being cooled to room temperature,the mixture was dilute with EtOAc (50 mL) and washed with water (5 mL × 3). The organic phase was dried over anhydrous Na2SO4,filtered and concentrated under reduced pressure. The crude oil was purified via flash column chromatography with EtOAc/petroleum ether (1:20) to give ketone 11 (38 mg,40%) as a colorless oil. 11: 1H NMR (400 MHz,CDCl3): δ 6.51 (t,1H,J = 4.8 Hz),6.38 (d,1H,J = 4.8 Hz),3.97 (d,1H,J = 7.2 Hz),3.81 (d,1H,J = 7.2 Hz),3.43 (s,1H),2.54 (d,1H,J = 12.4 Hz),2.44 (d,1H,J = 12.8 Hz),2.37 (dd,1H,J = 2.4,12.8 Hz),2.20 (d,1H,J = 12.4 Hz),0.88 (s,9H),0.09 (s,3H),0.08 (s,3H); 13C NMR (150 MHz,CDCl3): δ 208.09,207.85,134.60,131.29,61.70,56.73,49.56,37.39,36.32,25.77,18.19,-5.56. HRMS (ESITOF): m/z calcd. for (C16H26O3Si+H+): 295.1729; found 295.1728.
2.5. The preparation of compound 12To a stirred solution of the ketone 11 (30 mg,0.102 mmol) in THF (1 mL) was added a solution of TBAF (1.0 mol/l,0.5 mL,0.5 mmol) in THF,and the reaction mixture was stirred at room temperature for 2 h prior to being quenched with water (7 mL). The resulting mixture was extracted with EtOAc (15 mL × 3) and the combined organic extractswere dried over anhydrousNa2SO4,filtered. After removal of solvent in vacuo,the crude alcohol was dissolved in toluene (2 mL). To this solution were sequentially added ethylene glycol (19 mg,0.306 mmol) and p-TsOH (3 mg,0.0204 mmol),and the reaction mixture was heated at reflux for 1 h. The resultingmixture was concentrated in vacuo and purified by flash column chromatography with EtOAc/petroleum ether (1:5) to give ketal as a colorless oil. This oil was dissolved in dry CH2Cl2 (2 mL). To this solution was added Dess-Martin periodinane (67.6 mg,0.153 mmol),and the mixture was stirred for 2 h at 0 ℃ prior to being quenched by addition of saturated Na2S2O3 (4 mL). The mixture was extracted with CH2Cl2 (10 mL × 3). The organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified through flash chromatography with EtOAc/petroleum ether (1:5) to afford aldehyde 12 (10mg,47%) as a colorless oil. 12: 1H NMR (400 MHz,CDCl3): δ 9.91 (s,1H),6.61 (t,1H,J = 3.2 Hz),6.51 (d,1H,J = 5.6 Hz),4.03-3.94 (m,4H),2.97 (s,1H),2.62 (d,1H,J = 12.8 Hz),2.43 (d,1H,J = 9.6 Hz),2.13 (dd,1H,J = 2.4,12.4 Hz),2.02 (d,1H,J = 9.2 Hz); 13C NMR (150 MHz,CDCl3): δ 207.53,198.53,135.40,126.33,111.57,64.78,64.66,63.79,41.32,38.51,35.40. HRMS (ESI-TOF): m/z calcd. for (C15H24O3Si+Na+): 303.1392; found 303.1396.
2.6. The preparation of compound 13
To a stirred solution of 5 (105 mg,0.192 mmol) in dry toluene (3 mL) was slowly added a solution of t-BuOK (1.0 mol/L,0.144 mL) in dry THF under argon at 0 ℃,and the mixture was stirred at the same temperature for 30 min. A solution of 12 (20 mg,0.0961 mmol) in dry toluene (1 mL) was added dropwise to the above Wittig reagent,and the reaction mixture was stirred at 20 ℃ for 3 h. After being quenched by addition of saturated NH4Cl (5 mL),the resulting mixture was extracted with CH2Cl2 (10 mL × 3). The combined organic extracts were washed with brine,dried over anhydrous Na2SO4 and concentrated under vacuum to furnish a residue,which was subjected by flash column chromatography with EtOAc/petroleum ether (1:7) to give allene 13 (22 mg,66%) as a colorless oil. 13: 1H NMR (400 MHz,CDCl3): d 6.51-6.47 (m,1H),6.12-6.08 (m,1H),5.65 (d,0.5H,J = 7.6 Hz),5.54 (d,0.5H,J = 7.6 Hz),5.47-5.43 (m,1H),4.00-3.89 (m,8H),2.90 (s,1H),2.52 (d,1H,J = 12.0 Hz),2.37-2.25 (m,1H),2.17-2.04 (m,3H),1.83-1.80 (m,1H),1.74-1.67 (m,2H),1.=-1.43 (m,2H),1.32-1.21 (m,2H); 13C NMR (150 MHz,CDCl3): δ 208.12,208.04,139.33,139.10,135.29,135.06,134.09,134.01,124.66,124.57,112.26,112.16,108.56,108.48,64.58,64.53,
64.38,64.20,64.11,64.04,=.01,54.81,44.96,44.56,40.77,40.69,40.61,39.97,36.44,35.06,34.64,34,51,31.68,31.57,22.95,
22.90. HRMS (ESI-TOF):m/z calcd. for (C20H26O5+Na+): 369.1678; found 369.1674.
To a stirred solution of allene 13 (15 mg,0.0434 mmol) in THF (1 mL) was added 2 mol/L HCl (1 mL),and the mixture was heated at 60 ℃ for 8 h prior to be quenched by addition of saturated NaHCO3 (8 mL) at 0 ℃. The resulting mixture was extracted with EtOAc (10 mL × 3) and the combined organic extracts were washed with brine,dried over anhydrous Na2SO4 and concentrated to give a yellow residue. The residue was purified by flash chromatography with EtOAc/petroleum ether (1:5) to give a colorless oil. To a solution of the above oil in EtOAc (2 mL) was added Pd-C (3 mg),and the reaction mixture was stirred under an atmosphere of hydrogen at room temperature for 6 h. The mixture was filtered through Celite rinsing with EtOAc (15 mL × 3). The solvent was removed under reduced pressure to afford a residue,which was purified by flash column chromatography with EtOAc/ petroleum ether (1:6) to give ketone 14 (8 mg,68%) as a colorless oil. 14: 1H NMR(400 MHz,CDCl3): δ 2.74-2.73 (m,1H),2.51 (s,1H),2.45-2.43 (m,1H),2.39-2.34 (m,3H),2.28-2.22 (m,1H),2.05-1.99 (m,3H),1.96-1.91 (m,2H),1.81-1.75 (m,3H),1.65-1.51 (m,3H),1.31-1.27 (m,3H); 13C NMR (150 MHz,CDCl3): δ 211.82,211.77,211.56,211.45,49.05,48.03,44.85,44.25,44.04,41.45,40.73,39.52,31.03,30.60,30.15,27.97,27.73,25.07,
22.72. HRMS (ESITOF): m/z calcd. for (C16H22O3+Na+): 285.1467; found 285.1469.
To a stirred solution of ketone 14 (10 mg,0.0382 mmol) in anhydrous MeOH (1 mL) was added sodium methoxide (4 mg,0.0763 mmol) under argon,and the reaction mixture was heated at = ℃ for 10 h. After being cooled to 0 ℃,the mixture was extracted with EtOAc (6 mL × 3). The combined organic extracts were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography with EtOAc/petroleum ether (1:10) to give an unsaturated ketone 15 (5 mg,54%,diastereomeric ratio (d.r.) = 1:1) as a colorless oil. 15 upper: 1H NMR (400 MHz,CDCl3): δ 3.19 (d,1H,J = 13.2 Hz),2.66 (d,1H,J = 13.2 Hz),2.52 (dt,1H,J = 2.0,11.2 Hz),2.47 (t,1H,J = 2.4 Hz),2.37 (dd,1H,J = 1.6,12.4 Hz) 2.34-2.31 (m,1H),2.27-2.21 (m,1H),2.13 (d,1H,J = 12.4 Hz),1.98-1.89 (m,4H),1.79-1.66 (m,4H),1.48 (dt,1H,J = 1.6,9.2 Hz),1.36-1.27 (m,3H); 13C NMR (150 MHz,CDCl3): δ 215.32,202.42,150.70,131.93,48.97,43.56,42.19,39.01,38.60,33.82,33.53,32.20,30.81,27.42,22.75,22.51. 15 lower: 1H NMR (400 MHz,CDCl3): δ 3.07 (d,1H,J = 13.2 Hz),2.76 (dt,1H,J = 2.0,13.6 Hz),2.54 (dt,1H,J = 1.2,11.2 Hz),2.48 (s,1H),2.36- 2.26 (m,2H),2.21 (dd,1H,J = 2.0,12.8 Hz),2.17 (d,1H,J = 2.0 Hz),2.04-1.90 (m,4H),1.82-1.78 (m,2H),1.77-1.69 (m,2H),1.48 (dt,1H,J = 1.6,9.2 Hz),1.45-1.31 (m,3H). HRMS (ESI-TOF): m/z calcd. for (C16H20O2+Na+): 267.1361; found 267.1360.
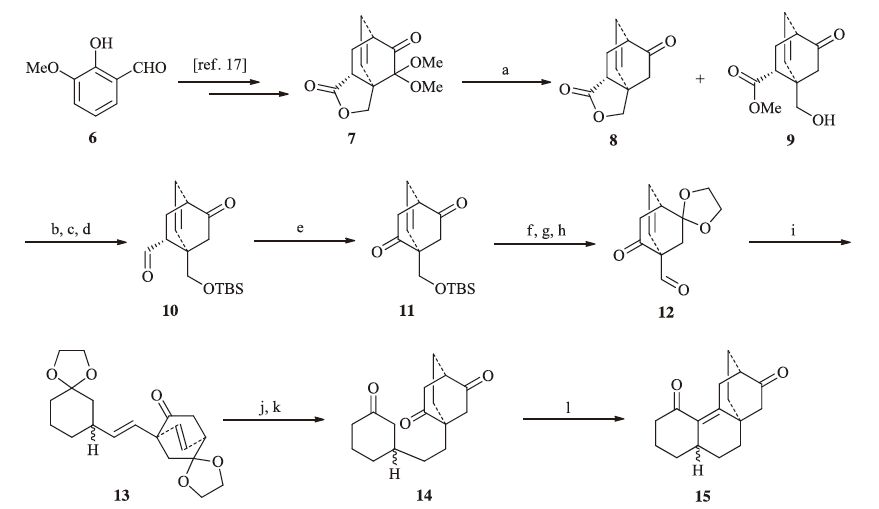
3. Results and discussionThe synthesis commenced with the preparation of the two building blocks,phosphonium salt 5 and aldehyde 12. The former arose from the known compound 4 [18],which was firstly iodinated under the standard condition (I2,Ph3P,imidazole),and then treated with Ph3P in toluene to furnish 5 in 52% overall yield (Scheme 1). On the other hand,the synthesis of the aldehyde segment started from our previous synthetic compound 7 (Scheme 2) [17]. Reductive deketalization of ketal 7 by treatment with SmI2 at room temperature gave the lactone 8 and alcohol 9 (1:3) in 95% overall yield [19]. Note that the latter was the lactone cleavage product,which could be used directly for the further transformations. Thus,silylation of the hydroxyl group in 9,followed by reduction of the carbonyl groups with DIBAL-H,and then oxidation of the resulting alcohols via Dess-Martin periodinane provided the aldehyde 10 (50% yield for three steps).

|
Download:
|
| Scheme 1.Synthesis of the phosphonium salt 5. Reagents and conditions: (a) I2,PPh3,imidazole,CH2Cl2,r.t.,3 h; (b) PPh3,tol,reflux,2 days,52% for two steps. | |
{kind=link}

|
Download:
|
| Scheme 2.Synthesis of the tetracyclic core of atisaneditepenes. Reagents and conditions: (a) SmI2,MeOH,THF,r.t.,30 min,95%; (b) TBSOTf,2,6-lutidine,CH2Cl2,r.t.,1 h; (c) DIBAL-H,CH2Cl2,0 ℃,2 h; (d) DMP,CH2Cl2,2 h,r.t.,50% for three steps; (e) O2,2,20-bipyridine,DABCO,Cu(OAc)2,DMF,70 ℃,6 h,40%; (f) TBAF,THF,r.t.,2 h; (g) ethylene glycol,p-TsOH,toluene,reflux,1 h; (h) DMP,CH2Cl2,0 ℃,2 h,47% for three steps; (i) 5,t-BuOK,toluene,0 ℃,3 h,66%; (j) 2moL/L HCl,THF,60 ℃,8 h; (k) H2,Pd-C,EtOAc,r.t.,6 h,68% for two steps; (l) NaOMe,MeOH,= ℃,54%. | |
{kind=link}
The next task was to get rid of the extra carbon to reach ketone 11. According to the method reported by Van Rheenen [20],the aldehyde 10 was exposed to O2 in the presence of 2,20-bipyridine,DABCO and cupric acetate in DMF,generating the desired product 11. However,after much experimentation,we found the transformation could only be achieved in moderate yield,while other methods gave poorer outcomes [21, 22]. After removal of the TBS group and protection of the carbonyl group,the resulting hydroxyl group was oxidated with Dess-Martin periodinane to afford aldehyde 12 (47% yield over three steps).
With the two segments in hand,we then investigated the Wittig reaction. Aldehyde 12 was treated with the Wittig reagent,generated in situ from phosphonium salt 5 and t-BuOK,to give diene 13 in 66% yield. Deprotection of 13 via treatment with acid,followed by global hydrogenation gave access to the ketone 14 in 68% overall yield. Finally,the intramolecular Aldol reaction took place smoothly under basic condition (NaOMe,MeOH) to furnish tetracyclic atisane framework 15 in 54% yield.
4. ConclusionIn summary,we described a convergent synthetic route to construct the tetracyclic core of atisane diterpenes. The readily available building blocks are assembled together via Wittig reaction. A ketone intermediate was then prepared through a sequence of deprotection/hydrogenation. A crucial intramolecular Aldol condensation process formed the a,β-unsaturated ketone. Further studies toward the total synthesis of atisane-type natural products are currently underway.
AcknowledgmentThis research was supported by the National Natural Science Foundation of China (No. 81273387).
| [1] | J.D. Connolly, R.A. Hill, Dictionary of Terpenoids, 1st. ed, Chapman and Hall, London, 1991. |
| [2] | J.R. Hanson, Diterpenoids, Nat. Prod. Rep. 17(2000) 165-174. |
| [3] | L.A. Mitscher, G.S.R. Rao, T. Veysoglu, S. Drake, T. Haas, Isolation and identification of trachyloban-19-oic and(-)-kaur-16-en-19-oic acids as antimicrobial agents from the prairie sunflower, helianthus annuus, J. Nat. Prod. 46(1983) 745-746. |
| [4] | S.Y. Ryu, J.W. Ahn, Y.N. Han, B.H. Han, S.H. Kim, In vitro antitumor activity of diterpenes from Aralia cordata, Arch. Pharm. Res. 19(1996) 77-78. |
| [5] | W.M. Daniewski, P. Skibicki, E. Bloszyk, M. Budesinsky, M. Holub, Constituents of Helianthus mollis lam. and their antifeedant activity, Polish J. Chem. 67(1993) 1255-1259. |
| [6] | M. Toyota, M. Yokota, M. Ihara, Construction of bicyclo[2.2.2] octane ring system via homoallyl-homoallyl radical rearrangement, Tetrahedron Lett. 40(1999) 1551-1554. |
| [7] | M. Ihara, M. Toyota, K. Fukuyama, T. Kametani, Stereocontrolled construction of a spiro fused bicyclo[2.2.2] Octane ring system by the intramolecular double michael reaction, Tetrahedron Lett. 25(1984) 2167-2170. |
| [8] | M. Ihara, M. Toyota, K. Fukuyama, T. Kametani, Intramolecular double Michael reaction. Part Ⅱ. Synthesis of isoatisirene type compound, Tetrahedron Lett. 25(1984) 3235-3238. |
| [9] | M. Ihara, M. Toyota, K. Fukuyama, T. Kametani, Intramolecular double Michael reaction ⅡI stereoselective chiral synthesis of atisiran-15-one, Tetrahedron Lett. 26(1985) 1537-1540. |
| [10] | M. Ihara, M. Toyota, K. Fukuyama, T. Kametani, An enantioselective total synthesis of(+)-atisirene by intramolecular double Michael reaction, J. Chem. Soc. Perkin Trans. 1(1986) 2151-2161. |
| [11] | M. Toyota, T. Wada, K. Fukuyama, M. Ihara, Total synthesis of(±)-methyl atis-16-en-19-oate via homoallyl-homoallyl radical rearrangement, J. Am. Chem. Soc. 120(1998) 4916-4925. |
| [12] | M. Toyota, T. Wada, M. Ihara, Total syntheses of(-)-methyl atis-16-en-19-oate,(-)-methyl kaur-16-en-19-oate, and(-)-methyl trachyloban-19-oate by a combination of palladium-catalyzed cycloalkenylation and homoallyl-homoallyl radical rearrangement, J. Org. Chem. 65(2000) 4565-4570. |
| [13] | E.C. Cherney, J.M. Lopchuk, J.C. Green, P.S. Baran, A unified approach to ent-atisane diterpenes and related alkaloids:synthesis of(-)-methyl atisenoate,(-)-isoatisine, and the hetidine skeleton, J. Am. Chem. Soc. 136(2014) 12592-12595. |
| [14] | M. Toyota, M. Yokota, M. Ihara, First total synthesis of(±)-methyl gummiferolate using a homoallyl-homoallyl radical rearrangement reaction, Org. Lett. 1(1999) 1627-1629. |
| [15] | M. Toyota, M. Yokota, M. Ihara, Remarkable control of radical cyclization processes of cyclic enyne:total syntheses of(±)-methyl gummiferolate,(±)-methyl 7β-hydroxykaurenoate, and(±)-methyl 7-oxokaurenoate and formal synthesis of(±)-gibberellin A12 from a common synthetic precursor, J. Am. Chem. Soc. 123(2001) 1856-1861. |
| [16] | M. Toyota, T. Asano, M. Ihara, Total synthesis of serofendic acids A and B employing tin-free homoallyl-homoallyl radicalrearrangement, Org. Lett.7(2005) 3929-3932. |
| [17] | D.L. Chen, F.P. Wang, Efficient synthesis of the C/D rings of atisine-type C20-diterpenoid alkaloids, Chin. Chem. Lett. 23(2012) 1378-1380. |
| [18] | F.A.J. Kerdesky, S.P. Schmidt, J.H. Holms, et al., Synthesis and 5-lipoxygenase inhibitory activity of 5-hydroperoxy-68, 11, 14-eicosatetraenoic acid analogs, J. Med. Chem. 30(1987) 1177-1186. |
| [19] | K.W. Tsao, B. Devendar, C.C. Liao, Ring rearrangement metathesis of 2-allylbicyclo[2.2.2] octenes:a short entry to cis-hydrindenols from 2-methoxyphenols, Tetrahedron Lett. 54(2013) 3055-3059. |
| [20] | V. Van Rheenen, Copper-catalyzed oxygenation of branched aldehydes-an efficient ketone synthesis, Tetrahedron Lett. 10(1969) 985-988. |
| [21] | J.A. Zallkowski, K.E. Gilbert, W.T. Borden, Oxidation of 7-(hydroxymethyl)bicyclo[3.3.1] nonan-3-ol. Convenient synthesis of bicyclo[3.3.1] nonane-3, 7-dione, J. Org. Chem. 45(1980) 346-347. |
| [22] | H.N. Sun, C. Yang, F. Gao, Z. Li, W.J. Xia, Oxidative C-C bond cleavage of aldehydes via visible-light photoredox catalysis, Org. Lett. 15(2013) 624-627. |