 2016, Vol.27
2016, Vol.27 


 , Zhi-Gang Zhao , Feng Chen
, Zhi-Gang Zhao , Feng Chen
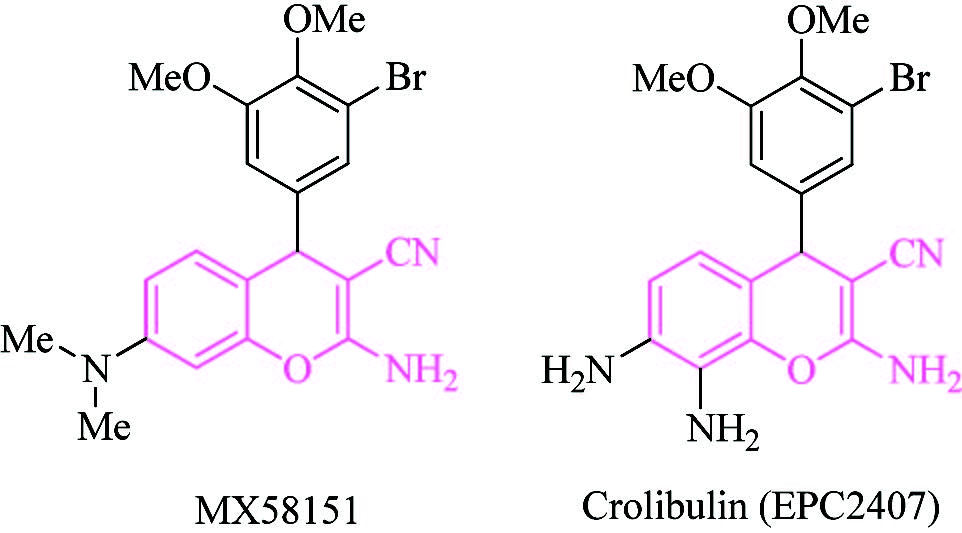
2-Amino-3-nitrile-chromene is an important medicinal scaffold and displays a wide range of biological properties. In addition to in vitro antibacterial activity [1],its derivatives might be employed to treat drug-resistant cancer. For example,compound MX58151 (Fig. 1) retained activity in tumor cells resistant towards current antimitotic agents,taxanes (including Taxol and Taxotere),and Vinca alkaloids [2]. Furthermore,these heterocycles were identified as vascular-disrupting agents (VDA),and one of the leading compounds,Crolibulin (EPC2407) (Fig. 1) [3],is currently in phase II clinical trials.

|
Download:
|
| Fig. 1.Structures of corresponding biologically active molecules. | |
Considering its attractive biological activities,the development of efficient synthetic approaches for this structure is of significant interest. Although there are numerous reports on the construction of its racemic form [4],examples of the catalytic asymmetric syntheses of these scaffolds were relatively less explored. Notably,the R-isomer of Crolibulin exhibited stronger antitumor activity (50-100 times more active) than the corresponding S-isomer [5]. Yang and Zhao successfully obtained 2-amino-3-nitrilechromene derivatives possessing a naphthene group via an addition-cyclization reaction of 2-naphthol with a,a-dicyanoolefins [6]. Later Xie et al. [7] and other groups [8] synthesized these heterocycles through the nucleophilic addition of malononitrile to a functional acceptor,followed by an intramolecular cyclization. Recently Yang and other groups developed a valuable catalytic process to synthesize enantiomerically pure 2-amino-4H-chromenes utilizing a Michael addition of 2-iminochromenes [9]. Although high yields and excellent enantioselectivity have been achieved in these published examples,itwas the modified organocatalysts rather than the natural products that exhibited optimal catalytic reactivity and stereoselectivity almost in all cases [10].
The synthesis of a suitable organocatalyst requires many redesigning sessions and long or expensive synthetic procedures. On the other hand,many enzymes,which are highly efficient biological catalysts,only displayed high activity and enantioselectivity when coenzymes were involved [11]. Motivated by the enzymatic systems,many readily available achiral additives were involved in the catalytic asymmetric transformations and an impressive improvement,in terms of reactivity and stereoselectivity was observed when compared with the use of organocatalysts alone [12]. This approach was beneficial in avoiding tedious chemical syntheses and would ultimately allow the highly efficient construction of libraries of catalyst systems by simply changing the additives. Based on our continuous efforts on asymmetric Michael reaction [13],herein we describe a domino process to construct 2- amino-3-nitrile-chromenes via a Michael-cyclization sequence of functionalized chalcones and malononitriles promoted by naturally occurring cinchona alkaloids.Notably,a remarkable enantioselectivity improvement was observed once additives were introduced to the catalyst system.
2. Experimental1H NMRand 13CNMRspectra were recorded on Varian 400 MHz spectrometers. ESI-HRMS spectra were recorded on a BioTOF instrument (Bruker Daltonics). Enantiomeric excess (ee) was determined by HPLC analysis on Chiralpak AS-H,AD-H,and ODH columns. Optical rotation data were recorded on an SGW-1 automatic polarimeter. The spectral data and spectra of all the compounds are presented in the Supporting information.
The functionalized chalcones 1a-q were prepared according to the procedures reported in literature [14]. Commercial grade solvents were dried and redistilled before use. All other reagents were purchased from commercial sources and used without further purification.
3. Results and discussionInitially,we examined the catalytic effect of a series of natural cinchona alkaloids. The designed cascade reaction of functional chalcone 1a and malononitrile proceeded smoothly in toluene and afforded the desired 2-amino-3-nitrile-chromene 3a,with quinine 4a (Fig. 2) giving an almost quantitative yield [15] (Table 1,entry 1). Cinchonidine 4b (Fig. 2) displayed lower catalytic activity and poorer enantioselectivity (entry 2). Quinidine 4c and cinchonine 4d (Fig. 2) delivered adducts with an opposite configuration and inferior optical purity than those delivered by quinine 4a (entries 3 and 4). We next studied the effect of the modified cinchona alkaloids. As we could see,the dihydroquinine 4e (Fig. 2) exhibited slightly higher enantioselectivity and poorer reactivity than quinine (entry 5). The C60-OH cinchona alkaloids 4f and 4g (Fig. 2) were examined and unsatisfactory stereoselectivity was detected in both cases (entries 6 and 7). A range of biscinchona alkaloids 4h-4k (Fig. 2) failed to complete the domino reaction even after one week,and less than 50% ee values were observed (entries 8-11). Furthermore,the 9-epi-amino cinchona alkaloid 4l and the corresponding thiourea 4m (Fig. 2) were investigated,and the desired products were obtained with marginal optical purity (entries 12 and 13). Moreover,when the Takemoto catalyst 4n (Fig. 2) was employed,only 36% ee was achieved (entry 14). The subsequent solvent screening revealed that polarity has a remarkable impact on the enantioselectivity (entries 15-20). Only marginal stereoselectivity was obtained when the more polar solvents,THF and MeOH,were used (entries 19 and 20). Unfortunately,no enantioselective improvement was observed when this transformation was performed at 0 ℃ (entry 21).
|
|
Table 1 Screening reaction conditions for the domino reaction of 1a with 2.a |

|
Download:
|
| Fig. 2.Structures of corresponding organocatalysts. | |
Using the identified quinine 4a as the optimal catalyst and toluene as the solvent of choice,we next focused on effects of the additives on the enantioselectivity and catalytic activity. The addition of molecular sieves (M.S.,4A˚ ) led to an impairment of the reaction rate but negligible improvement of ee values (Table 2,entries 1 and 2). It has been documented that the structural modifications of cinchona alkaloids exerted a direct impact on their asymmetric induction [16]. As previously reported,acids might protonate the N-quinuclidine moiety and induce conformational changes of cinchona alkaloids,resulting in different catalytic behaviors [17]. Inspired by Baiker’s observations on the heterogeneous hydrogenation of ethyl pyruvate [17b,c],we attempted to introduce readily available acidic additives to the catalytic system. As summarized in Table 2,when 5 mol% of aromatic carboxylic acid was added,a slight enantioselectivity increase was attained at the cost of a decrease in reactivity (entries 3-6). Aliphatic carboxylic acids and sulfonic acids were also examined (entries 7-9) and acetic acid enhanced the ee value from an original value of 68-76% (entry 7). Considering the strong acidity of carboxylic acids,in the following study we performed the tandem reaction of 1a with malononitrile in the presence of less acidic phenolic compounds (entries 10-23) [12e,h]. When one equivalent of phenol and its analogues were added in the cascade reaction,a significant decrease in reactivity was observed and prolonged reaction time was required in almost all cases. The use of phenolic compounds improved the enantioselectivity to a greater extent when compared to carboxylic and sulfonic acids (entries 10-23 vs. 3-9). The electronic effect on the aromatic ring of phenol was not apparent (entries 11-14 vs. 10). The electron-rich para-methyl substituted phenol afforded 75% ee (entry 11) while the electronpoor 2,4,6-trinitrophenol delivered 78% ee (entry 14). Further studies indicated that a-naphthol greatly improved the enantioselectivity and 81% ee was detected (entry 15). However,β-naphthol only generated the desired compound with 74% ee (entry 16). Considering the poor conversion of a-naphthol,the subsequent investigation focused on the loading of additive. A decrease in loading led to a significant conversion enhancement (entries 17-20). The transformation completed within 72 h in the presence of 20 or 30 mol% a-naphthol,however,the enantioselectivity also reduced (entries 17 and 18). Fortunately,the enantioselectivity remained in the presence of 50 mol% a-naphthol and almost quantitative yield was obtained after one week (entry 20). Next,we added a-naphthol together with the molecular sieves to the reaction. This resulted in an inferior enantioselectivity (76% ee) (entry 21). The chiral additive 1,10-bi-2-naphthol (BINOL) was also evaluated [12g] (entries 22 and 23) and poorer stereo control was observed for both additives. Both (R)-BINOL and (S)-BINOL afforded the desired heterocycles with the same configuration. It further confirmed that it’s the quinine rather than the additives that played a crucial role during the course of asymmetric induction. Finally,alcohols were found to slightly enhance the enantioselectivity (entries 24 and 25).
|
|
Table 2 Additives screening for the domino reaction of 1a with 2.a |

Having established the optimal reaction conditions,we subsequently examined the substrate scope of the domino reaction of functionalized chalcone 1 and malononitrile 2. As summarized in Table 3,all the reaction proceeded smoothly in the presence of 20 mol% quinine and 50 mol% a-naphthol in toluene and the desired chromenes were generated in high yields (75%-99%) and moderate to good enantioselectivity (49%-84% ee). The substrate scope study revealed that electron-donating and electron-withdrawing groups,locating at the phenyl group adjacent to the carbonyl group,were all well tolerated. All the substrates 1b-g underwent efficient cascade reactions with malononitrile,affording the desired heterocycles in high yields (entries 2-7). Generally speaking,the substrates containing electron-withdrawing groups afforded comparable ee values with the substrates containing electron-donating groups (entries 5-7 vs. 3-4). An ortho-substituted chalcone 1b produced the corresponding product with poorer enantioselectivity (49% ee),probably caused by the steric effect of the methyl group (entry 2). Moreover,reactions with heteroaryl-containing substrates 1h-i proceeded well and the desired products were obtained with moderate stereoselectivity (entries 8 and 9). Notably,a pronounced positive additive effect was observed again in 1b-i when the a-naphthol was involved under otherwise identical conditions (entries 2-9,data outside brackets vs. data in brackets). Up to 37% enantioselectivity improvement was observed in the case of 1i with a thienyl group (entry 9). We next investigated the effect of another phenyl group on the terminal double bond in the functionalized chalcones (entries 10-12). Compounds 1j-l successfully converted into the desired chromenes. Almost quantitative yields and better enantioselectivity could be achieved at 10 ℃ when compared to reactions conducted at 23 ℃. Gratifyingly,up to 84% ee was achieved for substrate 1l with two bromine atoms on the phenyl group (entry 12). Further studies indicated that good enantioselectivity remained for dibromo-substituted chalcones 1m-q when electron-donating and electron-withdrawing groups were introduced to the phenyl group adjacent to the carbonyl group (entries 13-17). The configuration of the products was determined to be R on the basis of the reported optical rotation. [7]
|
|
Table 3 Substrate spectrum investigation of the domino reaction of 1 and 2.a |

{kind=link}
{kind=link}
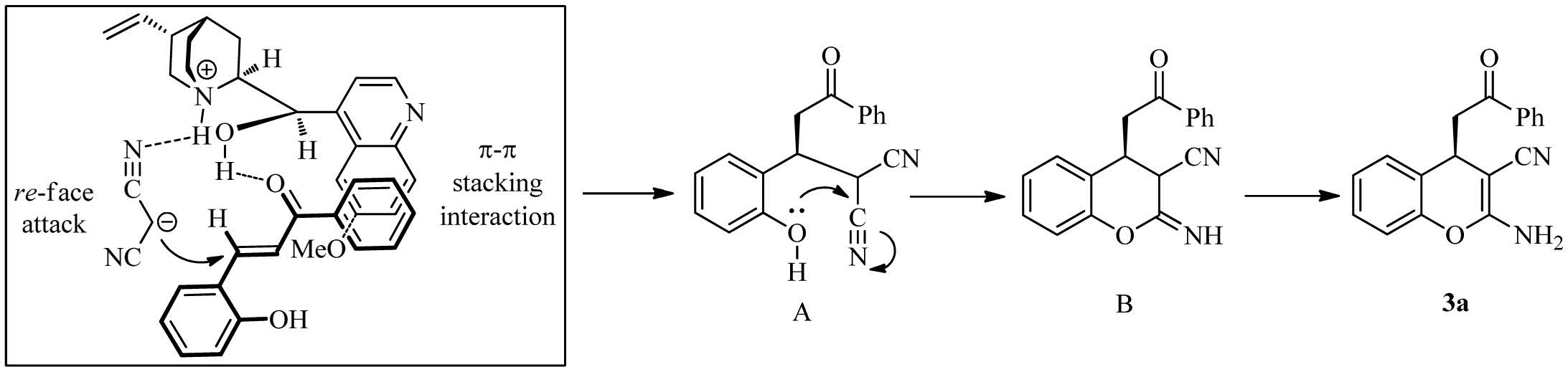
The postulated reaction pathway is summarized in Scheme1.Our domino process might share a similar transition state as Lattanzi’s enantioselective Michael reaction [15] due to the similar configuration of the product. In the initial step,malononitrile was deprotonated by the tertiary aminemoiety of quinine and orientated via a hydrogen bond. Meanwhile,the hydroxy groupwas involved in a hydrogen-bonding interaction with the oxygen in the carbonyl moiety inchalcone1a. Theformationof a ternarycomplex of quinine,chalcone 1a,and malononitrile would stereoselectively afford intermediate A. Subsequently,the intermolecular oxo-nucleophilic addition of the nitrile group led to intermediate B. Finally,intermediate C underwent tautomerization to give the desired compound 3a. The preferential attack of malononitrile towards the re-face of the enone might likely be assisted by a favorable π-π stacking interaction between the quinoline residue of the catalyst and the aromatic moiety linked to the carbonyl group. This resulted in an R-configured product.
To further investigate the role of additive,the Michael addition of an ortho-methoxy substituted chalcone 5 with malononitrile was conducted under the identical conditions mentioned above (Scheme 2). Notably,the beneficial effect was observed again. The process generated the desired adduct with 79% ee in the absence of a-naphthol; however,up to 82% ee was obtained once 50 mol% anaphthol was introduced. This confirmed that the additive effect was general. The participation of a-naphthol resulted in a decrease in the reaction rate and an improvement in enantioselectivity (Table 1,entry 1 vs. Table 2,entry 20; Table 3,entries 2-9). We postulated that the acidic a-naphthol could attach to quinine via the protonation of the tertiary amine moiety of quinine. This would disturb the deprotonation of malononitrile and slow down the reaction rate. At the same time,the resulting complex of quinine and a-naphthol might possess catalytic activity and afforded better enantioselectivity when compared to the process solely catalyzed by quinine [16]. As a result,a significant enhancement of enantioselectivity was observed. Nevertheless,the real catalytic mechanism still needs further investigation.

|
Download:
|
| Scheme 1.Proposed mechanism for the domino reaction. | |
{kind=link}

|
Download:
|
| Scheme 2.Michael addition of an ortho-methoxy substituted chalcone 5 with malononitrile | |
{kind=link}
We have developed an efficient strategy to construct 2-amino- 3-nitrile-chromenes,with potential antitumor activity,from readily accessible functionalized chalcones and malononitrile. The natural cinchona alkaloids such as quinine could efficiently promote the tandem conversion to produce the desired heterocycles in high yields (75%-99%) and moderate to good enantioselectivity (49%-84% ee). In particular,achiral a-naphthol exerted a positive impact to the stereoselectivity and a significant improvement in enantioselectivity. The beneficial additive effect was frequently observed in the case of primary and secondary aminepromoted transformations; however,it was seldom found in the process catalyzed by tertiary amines. Although an enhancement of reactivity and enantioselectivity was observed in the case of the heterogeneous catalytic enantioselective hydrogenation of ethyl pyruvate after achiral tertiary amines were utilized,the cinchona alkaloids were employed in the reaction as the ligand and not the organocatalyst [18]. Further research actively performed in our lab concentrates on clarifying the definite role of a-naphthol and the synthetic utilizations of the attained product.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (No. 21402163) and the Science and Technology Department of Sichuan Province (No. 2013JY0135). Zheng Yuanqin gratefully acknowledges the Graduate Innovation Project of Southwest University for Nationalities (No. CX2015SZ064).
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.08.013.
| [1] | (a) M. Kidwai, S. Saxena, M.K. Khan, et al., Aqua mediated synthesis of substituted 2-amino-4H-chromenes and in vitro study as antibacterial agents, Bioorg. Med. Chem. Lett. 15(2005) 4295-4298;(b) N.J. Thumar, M.P. Patel, Synthesis and in vitro antimicrobial evaluation of 4Hpyrazolopyran-benzopyran and naphthopyran derivatives of 1H-pyrazole, Arkivoc(2009) 363-380;(c) N.M. Sabry, H.M. Mohamed, E.S. Khattab, et al., Synthesis of 4H-chromene, coumarin, 12H-chromeno[2,3-d]pyrimidine derivatives and some of their antimicrobial and cytotoxicity activities, Eur J. Med. Chem. 46(2011) 765-772. |
| [2] | (a) S. Kasibhatla, H. Gourdeau, K. Meerovitch, et al., Discovery and mechanism of action of a novel series of apoptosis inducers with potential vascular targeting activity, Mol. Cancer Ther. 3(2004) 1365-1374;(b) H. Gourdeau, L. Leblond, B. Hamelin, et al., Antivascular and antitumor evaluation of 2-amino-4-(3-bromo-4, 5-dimethoxy-phenyl)-3-cyano-4H-chromenes, a novel series of anticancer agents,, Mol. Cancer Ther. 3(2004) 1375-1384. |
| [3] | (a) S.X. Cai, Small molecule vascular disrupting agents:potential new drugs for cancer treatment, Recent Pat. Anticancer Drug Discov. 2(2007) 79-101;(b) S.X. Cai, J. Drewe, W. Kemnitzer, Discovery of 4-aryl-4H-chromenes as potent apoptosis inducers using a cell-and caspase-based anti-cancer screening apoptosis program(ASAP):SAR studies and the identification of novel vascular disrupting agents, Anticancer Agents Med. Chem. 9(2009) 437-456. |
| [4] | (a) G. Yin, H. Shi, L. Xu, et al., Selective synthesis of cyano-functionalized 2-aryl-4H-chromenes and 2-amino-4H-chromene-3-carbonitriles by catalyst-tuned reactions of 2-hydroxychalcones with 2-substituted acetonitriles, Synthesis 45(2013) 334-340;(b) H.F. Gan, W.W. Cao, Z. Fang, et al., Efficient synthesis of chromenopyridine and chromene via MCRs, Chin. Chem. Lett. 25(2014) 1357-1362;(c) M.A. Ameen, S.M. Motamed, F.F. Abdel-latif, Highly efficient one-pot synthesis of dihydropyran heterocycles, Chin. Chem. Lett. 25(2014) 212-214;(d) J. Albadi, A. Mansournezhad, M. Darvishi-Paduk, Poly(4-vinylpyridine):as a green, efficient and commercial available basic catalyst for the synthesis of chromene derivatives, Chin. Chem. Lett. 24(2013) 208-210. |
| [5] | (a) S.X. Cai, J.A. Drewe, S. Kasibhatla, et al., Substituted 4-aryl-chromene as activator of caspases and inducer of apoptosis and as antivascular agent and the use thereof, U.S. Patent and Trademark Office, Washington, DC, 2011(Patent No.:7,968,595 B2);(b) A.M. Shestopalov, Y.M. Litvinov, L.A. Rodinovskaya, et al., Polyalkoxy substituted 4H-chromenes:synthesis by domino reaction and anticancer activity, ACS Comb. Sci. 14(2012) 484-490. |
| [6] | X.S. Wang, G.S. Yang, G. Zhao, Enantioselective synthesis of naphthopyran derivatives catalyzed by bifunctional thiourea-tertiary amines, Tetrahedron:Asymmetry 19(2008) 709-714. |
| [7] | J.W. Xie, X. Huang, L.P. Fan, et al., Efficient method for the synthesis of optically active 2-amino-2-chromene derivatives via one-pot tandem reactions, Adv. Synth. Catal. 351(2009) 3077-3082. |
| [8] | (a) Y. Gao, W. Yang, D.M. Du, Efficient organocatalytic asymmetric synthesis of 2-amino-4H-chromene-3-carbonitrile derivatives, Tetrahedron:Asymmetry 23(2012) 339-344;(b) K. Hu, A. Lu, Y. Wang, et al., Chiral bifunctional squaramide catalyzed asymmetric tandem Michael-cyclization reaction:efficient synthesis of optically active 2-amino-4H-chromene-3-carbonitrile derivatives, Tetrahedron:Asymmetry 24(2013) 953-957;(c) Q. Ren, W.Y. Siau, Z. Du, et al., Expeditious assembly of a 2-amino-4Hchromene skeleton by using an enantioselective Mannich intramolecular ring cyclization-tautomerization cascade sequence, Chem. Eur. J. 17(2011) 7781-7785. |
| [9] | (a) G. Yang, C. Luo, X. Mu, et al., Highly efficient enantioselective three-component synthesis of 2-amino-4H-chromenes catalysed by chiral tertiary aminethioureas, Chem. Commun. 48(2012) 5880-5882;(b) W. Li, J. Huang, J. Wang, Organocatalytic conjugate addition promoted by multi-hydrogen-bond cooperation:access to chiral 2-amino-3-nitrile-chromenes, Org. Biomol. Chem. 11(2013) 400-406;(c) W. Li, H. Liu, X. Jiang, et al., Enantioselective organocatalytic conjugate addition of nitroalkanes to electrophilic 2-iminochromenes, ACS Catal. 2(2012) 1535-1538;(d) Y. Gao, D.M. Du, Facile synthesis of chiral 2-amino-4-(indol-3-yl)-4H-chromene derivatives using thiourea as the catalyst, Tetrahedron:Asymmetry 24(2013) 1312-1317;(e) W. Chen, Y. Cai, X. Fu, et al., Enantioselective one-pot synthesis of 2-amino-4-(indol-3-yl)-4H-chromenes, Org. Lett. 13(2011) 4910-4913. |
| [10] | A. Adili, Z.L. Tao, D.F. Chen, et al., Quinine-catalyzed highly enantioselective cycloannulation of o-quinone methides with malononitrile, Org. Biomol. Chem. 13(2015) 2247-2250. |
| [11] | (a) C.H. Wong, L. Daniels, W.H. Orme-Johnson, et al., Enzyme-catalyzed organic synthesis:NAD(P)H regeneration using dihydrogen and the hydrogenase from Metbanobacterium tbermoautotropbicum, J. Am. Chem. Soc. 103(1981) 6227-6228;(b) G.Hambreus,N.Nyberg,Enzymatichydrogenationoftrans-2-nonenalinbarley, J. Agric. Food Chem. 53(2005) 8714-8721. |
| [12] | (a) E.M. Vogl, H. Gröger, M. Shibasaki, Towards perfect asymmetric catalysis:additives and cocatalysts, Angew. Chem. Int. Ed. 38(1999) 1570-1577;(b) A. Martinez-Castaneda, B. Poladura, H. Rodriguez-Solla, et al., Highly enantioselective proline-catalysed direct aldol reaction of chloroacetone and aromatic aldehydes, Chem. Eur. J. 18(2012) 5188-5190;(c) A. Martinez-Castaneda, K. Kedziora, I. Lavandera, et al., Highly enantioselective synthesis of alpha-azido-beta-hydroxy methyl ketones catalyzed by a cooperative proline-guanidinium salt system, Chem. Commun. 50(2014) 2598-2600;(d) A. Martinez-Castaneda, H. Rodriguez-Solla, C. Concellon, et al., Switching diastereoselectivity in proline-catalyzed aldol reactions, J. Org. Chem. 77(2012) 10375-10381;(e) T.J. Peelen, Y. Chi, S.H. Gellman, Enantioselective organocatalytic Michael additions of aldehydes to enones with imidazolidinones:cocatalyst effects and evidence for an enamine intermediate, J. Am. Chem. Soc. 127(2005) 11598-11599;(f) S. Kuwano, S. Harada, B. Kang, et al., Enhanced rate and selectivity by carboxylate salt as a basic cocatalyst in chiral N-heterocyclic carbene-catalyzed asymmetric acylation of secondary alcohols, J. Am. Chem. Soc. 135(2013) 11485-11488;(g) Y. Zhou, Z. Shan, Chiral diols:a new class of additives for direct aldol reaction catalyzed by L-proline, J. Org. Chem. 71(2006) 9510-9512;(h) C.S. Da, L.P. Che, Q.P. Guo, et al., 2, 4-Dinitrophenol as an effective cocatalyst:greatly improving the activities and enantioselectivities of primary amine organocatalysts for asymmetric aldol reactions,, J. Org. Chem. 74(2009) 2541-2546;(i) A.N. Martínez-Castañeda, B. Poladura, H. Rodríguez-Solla, et al., Direct aldol reactions catalyzed by a heterogeneous guanidinium salt/proline system under solvent-free conditions, Org. Lett. 13(2011) 3032-3035. |
| [13] | (a) X. Li, L. Cun, C. Lian, et al., Highly enantioselective Michael addition of malononitrile to α,β-unsaturated ketones, Org. Biomol. Chem. 6(2008) 349-353;(b) X. Li, Y. Ma, Z. Xing, et al., The asymmetric addition of malononitrile to α,β-unsaturated ketones catalyzed by RuCl2[(R,R)-DPEN](PPh3)2 as the precatalyst, Tetrahedron Lett. 55(2014) 3868-3872. |
| [14] | G. Yin, L. Fan, T. Ren, et al., Synthesis of functionalized 2-aryl-4-(indol-3-yl)-4Hchromenes via iodine-catalyzed domino Michael addition-intramolecular cyclization reaction, Org. Biomol. Chem. 10(2012) 8877-8883. |
| [15] | A. Russo, A. Perfetto, A. Lattanzi, Back to natural cinchona alkaloids:highly enantioselective Michael addition of malononitrile to enones, Adv. Synth. Catal. 351(2009) 3067-3071. |
| [16] | (a) P. Melchiorre, Cinchona-based primary amine catalysis in the asymmetric functionalization of carbonyl compounds, Angew. Chem. Int. Ed. 51(2012) 9748-9770;(b) G.D. Dijkstra, R.M. Kellogg, H. Wynberg, et al., Conformational study of cinchona alkaloids. A combined NMR, molecular mechanics and x-ray approach,, J. Am. Chem. Soc. 111(1989) 8069-8076;(c) T. Bürgi, A. Baiker, Conformational behavior of cinchonidine in different solvents:a combined NMR and ab initio investigation,J.Am.Chem.Soc.120(1998)12920-12926;(d) A.Urakawa,D.M.Meier,H.Ruègger,etal.,Conformational behavior of cinchonidine revisited:a combined theoretical and experimental study, J. Phys. Chem. A 112(2008) 7250-7255. |
| [17] | (a) R.A. Olsen, D. Borchardt, L. Mink, et al., Effect of protonation on the conformation of cinchonidine, J. Am. Chem. Soc. 128(2006) 15594-15595;(b) B. Minder, T. Mallat, P. Skrabal, et al., Enantioselective hydrogenation of ethyl pyruvate. Influenceof oxidative treatment of cinchonidine-modifiedplatinum catalyst and hemiketal formation in alcoholic solvents, Catal. Lett. 29(1994) 115-124;(c) D.Ferri,T.Bürgi,K.Borszeky,etal.,Enhanced enantioselectivity in ethyl pyruvate hydrogenation due to competing enantioselective aldolreaction catalyzed by cinchonidine, J. Catal. 193(2000) 139-144. |
| [18] | J.L. Margitfalvi, E. Tálas, F. Zsila, et al., Dimer formation of cinchonidine in liquid phase:relevance to the heterogeneous catalytic enantioselective hydrogenation of ethyl pyruvate, Tetrahedron:Asymmetry 18(2007) 750-758. |