 2015, Vol.26
2015, Vol.26 



b Fragrance Ingredient Research Center, Givaudan Fragrances (Shanghai) Ltd., Shanghai 201203, China
Since the first isolation of pentadecanolide from angelica oil in 1927 by Kerschbaum,the interest in synthesis and properties of macrocyclic lactones has constantly grown [1]. Macrolides are ubiquitous motifs in natural products,pharmaceuticals and odorants [2]. The widespread occurrence in biologically active natural products and therapeutic agents makes the development of efficient strategies for their assembly a longstanding topic in organic synthesis. Even though a variety of efficient lactonization methods such as the ring-closing metathesis (RCM),intramolecular cross-coupling such as the Corey-Nicolaou S-pyridyl ester lactonization method,the Yamaguchi mixed-anhydride method, the Mitsunobu alcohol activation method or HWE methods have been developed over the past decades,there remains a high demand for mild,atom-economical new strategies [3]. In particular, Z-selective macrocyclization methods are sought after. Quite recently,Kananovich and coworkers have reported the synthesis of medium-ring-sized trans-alkenolides via PhI(OCOCF3)2-mediated oxidative fragmentation of bicyclo[n.1.0]alkan-1-ols and the application to the synthesis of (+)-recifeiolide. However,the precursors require a multi-step synthesis [4].
We have recently developed an unprecedented oxy-oxonia (azonia)-Cope rearrangements based on a tandem Lewis acidcatalyzed crossed dimerization of β,γ-unsaturated carbonyl compounds with other different aldehydes (imines) to produce homoallylic esters (amides) via disproportionation [5]. The reaction has been successfully extended to a novel [n + 4] ring enlargement toward macrolides with 9- to 16-membered rings. However,when we tried to apply this strategy to synthesize some natural products with simple internal double bonds such as recifeiolide [4, 6],we faced limitations with regard to scope of substrate: the β,γ-unsaturated ketones we used before predominantly carried a β-substituent,such as a methyl group at the double bond. This was a beneficial structural feature to stabilize the involved zwitterionic intermediates. Of course,the products from the [n + 4] ring enlargement rearrangement would also have that substituent incorporated at the newly formed double bond which would not allow efficient syntheses of less substituted target molecules. Therefore,we intended to introduce removable group as a temporally masked proton which could also deliver the required stabilization aspect. It has been well documented that silicon-based protecting groups have a pronounced tendency to stabilize adjacent positive charges [7] and carbocations bearing an α-silicon group are more stable than their carbon analogs. We anticipated that a β-silicon-substitution on the β,γ-unsaturated substrate would be capable of stabilizing the zwitterionic intermediate and thereby facilitate the reaction. Moreover, silicon-carbon bonds can be easily cleaved which offers a solution to the above mentioned substitution problem. In this regard,we report herein a highly efficient synthesis of (±)-Z-recifeiolide (6) based on an oxy-oxonia-Cope rearrangement [8, 9, 10]. In comparison to other methods,the present strategy,outlined in Scheme 1, represents a novel example of a synthetic pathway to Z-configured 12-membered alkenolides.

|
Download:
|
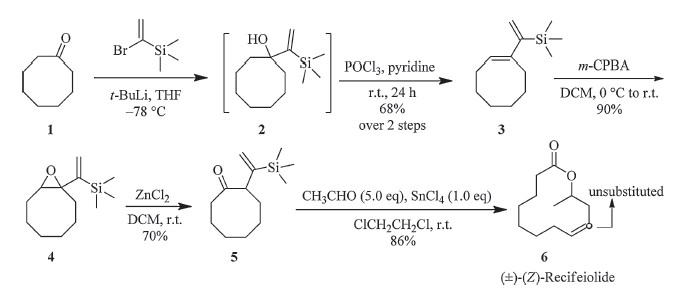
| Scheme. 1.The synthesis of (±)-Z-recifeiolide (6). | |
{kind=link}
All reactions were performed under argon using solvents and reagents from commercial suppliers without further purification. Solvents for extraction and chromatography were technical grade and used without further purification. Flash chromatography was performed using Tsingdao Haiyang Chemical silica gel (200 ± 300 mesh) and silica gel Merck grade (60Å ). Unless otherwise noted,a mixture of hexane:MTBE (20:1) was used as eluent. NMR spectra were recorded with an AW 300 MHz Bruker spectrometer instrument. The chemical shifts for 1H NMR spectra are reported in d (ppm) referenced to the residual proton signal of the deuterated solvent; coupling constants are expressed in Hertz (Hz). 13C NMR spectra were referenced to the carbon signals of the deuterated solvent; the nature of the carbons (C,CH,CH2,or CH3) was determined by recording the DEPT-90 and DEPT-135 experiments. IR spectra were recorded with a Bruker Tensor 27 and a Jasco FT/IR-4100. High resolution MS were obtained with a Bruker microTOF II spectrometer (ESI-MS). The applied acetaldehyde was an Aldrich product with 99% purity and kept in cool.
Preparation of 1-(1-trimethylsilyl)vinylcyclooctene (3): [2TD$DIF]To a solution of (α-bromovinyl)trimethylsilane (9.00 g,50.0 mmol) in dry THF (50 mL) at -78 ℃ under argon was added drop wise a solution of tert-butyllithium (1.3 mol/L in pentane,50 mL, 65 mmol,Alfa Aesar Product). After completion of the addition, the mixture was stirred at -78 ℃ for an additional 60 min. Then a solution of cyclooctanone 1 (5.05 g,40.0 mmol) in dry THF (40 mL) was added dropwise with stirring. The mixture was allowed to slowly warm to room temperature overnight. The reaction was cooled,quenched with saturated NH4Cl solution (100 mL),and extracted thoroughly with ether (100 mL × 3). The combined ether fractions were washed,dried,and concentrated at reduced pressure to give the 1-vinyl cyclooctanol 2. Without further purification,the crude alcohol 2 was dissolved in four times its volume of pyridine. To the resultant solution 10% excess of POCl3 were added slowly at 0-5 ℃ under argon with stirring. After 24 h of stirring at room temperature the mixture was poured into icy water. The mixture was extracted with hexane,washed,dried, concentrated at reduced pressure,and purified by column chromatography on silica gel to give compound 3 (5.70 g, 27.4 mmol,68% yield over two steps) as a colorless liquid. The freshly prepared diene product 3 was sealed under argon and stored in the refrigerator to prevent polymerization prior to use. IR (neat,cm-1): v 2923,2850,1448,1247,914,833,758. 1H NMR (CDCl3,300 MHz): δ 5.64 (d,1H,J = 2.7 Hz),5.58 (t,1H,J = 6.5 Hz), 5.33 (d,1H,J = 2.7 Hz),2.37 (t,2H,J = 7.5 Hz),2.30-2.12 (m,2H), 1.62-1.35 (m,8H),0.16 (s,9H). 13C NMR (CDCl3,75 MHz): δ 152.9 (s),143.0 (s),128.6 (d),123.2 (t),30.5 (t),28.7 (t),27.5 (t),27.2 (t), 26.6 (t),26.3 (t),0.1 (q). HRMS (ESI) (m/z) calcd. for C13H24Si + H+: 209.1726; found: 209.1724.
Preparation of 9-oxa-1-(1-trimethylsilyl)vinyl-bicyclo[6.1.0]- nonane (4):m-CPBA (5.18 g,w/w% 85%,25.5 mmol) was added to a solution of 3 (5.20 g,25.0 mmol) in CH2Cl2 (50 mL) at 0 ℃ and the mixture was stirred at this temperature for 1 h. The reaction was quenched with NaOH (10%) and the aqueous layer was extracted with CH2Cl2 (50 mL × 2). The organic extracts were dried over MgSO4 and concentrated under reduced pressure to give the crude oily product,which was further purified by flash chromatography on silica gel to furnish compound 4 (5.00 g,22.3 mmol,yield 90%) as a colorless liquid. IR (neat,cm-1): v 2929,1469,1246,936,836, 762. 1H NMR (CDCl3,300 MHz): δ 5.80 (d,1H,J = 2.4 Hz),5.55 (d, 1H,J = 2.4 Hz),2.93 (dd,1H,J = 9.9,4.5 Hz),2.33-2.13 (m,1H), 2.07-1.94 (m,1H),1.66-1.35 (m,10H),0.15 (s,9H). 13C NMR (CDCl3,75 MHz): δ 152.2 (s),127.2 (t),67.0 (s),61.6 (d),30.9 (t), 28.1 (t),26.6 (t),26.6 (t),25.4 (t),25.4 (t),-0.2 (q). HRMS (ESI) (m/z) calcd. for C13H24OSi + H+: 225.1675; found: 225.1679.
Preparation of 2-(1-(trimethylsilyl)vinyl)cyclooctanone (5): Anhydrous zinc chloride (0.27 g,2.0 mmol) was added to a solution of 4 (4.50 g,20.0 mmol) in CH2Cl2 (40 mL) at room temperature under argon with stirring. The mixture was stirred at this temperature for an additional 1 h. The reaction was quenched with sat. aqueous NaHCO3 solution (40 mL) and the aqueous layer was extracted with MTBE (50 mL × 3). The organic extracts were dried over MgSO4 and evaporated in vacuo. The residue was purified by flash column chromatography on silica gel to give ketone 5 (3.14 g,yield 70%) as a colorless liquid. IR (neat,cm-1): v 2928,1696,1466,1247,927,835. 1H NMR (CDCl3,300 MHz): δ 5.83 (d,1H,J = 1.7 Hz),5.51 (d,1H,J = 1.7 Hz),3.30 (d,1H, J = 11.4 Hz),2.25-2.00 (m,2H),1.91-1.37 (m,10H),0.08 (s,9H). 13C NMR (CDCl3,75 MHz): δ 217.0 (s),150.8 (s),125.9 (t),56.4 (d),39.8 (t),31.0 (t),27.5 (t),27.2 (t),26.7 (t),24.8 (t),-1.1 (q). HRMS (ESI) (m/z) calcd. for C13H24OSi + H+: 225.1675; found: 225.1675.
Preparation of (Z)-8-dodecen-11-olide ((Z)-Recifeiolide) (6): [4TD$DIF]An argon flushed three-necked flask was charged with the above b,gunsaturated ketone 5 (0.45 g,2.0 mmol),acetaldehyde (0.45 g, 10.0 mmol) and 1,2-dichloroethane (5.0 mL). Anhydrous stannic chloride (0.52 g,2.0 mmol) was added dropwise under argon at room temperature. After completion of the addition,the mixture was stirred for 10 h at room temperature. The progress of the reaction was monitored by GC analysis of reaction aliquots quenched with a solution of saturated NaHCO3 in water. After full conversion,the reaction mixture was quenched with sat. aqueous NaHCO3 solution (10 mL). The organic phase was separated and the aqueous layer was extracted with MTBE (10 mL × 2). The combined organic layers were washed with brine (20 mL),dried (MgSO4) and evaporated in vacuo. The residue was purified by column chromatography on silica gel (MTBE/ hexane = 1:20) to yield (Z)-recifeiolide 6 as a colorless liquid (0.34 g,86% yield). IR (neat,cm-1): v 2930,2857,1727,1448,1361, 1222,1190,1152,1132,1044,707. 1H NMR (CDCl3,300 MHz): δ 5.63-5.36 (m,2H),5.34-5.16 (m,1H),2.57-2.12 (m,5H),2.07-1.95 (m,1H),1.90-1.76 (m,1H),1.63-1.35 (m,5H),1.29 (d,3H, J = 6.6 Hz),1.24-1.09 (m,1H). 13C NMR (CDCl3,75 MHz): δ 173.3 (s),133.5 (d),124.0 (d),69.0 (d),33.1 (t),32.7 (t),27.1 (t),25.1 (t), 25.0 (t),24.3 (t),24.3 (t),19.20 (q). HRMS (ESI) (m/z) calcd. for C12H20O2 + H+: 197.1542; found: 197.1540.
3. Results and discussionOur study began with the preparation of the required key material 5. As illustrated in Scheme 1,it was easily accessible in four steps. The in situ prepared organolithium reagent was allowed to react with cyclooctanone 1 to afford the 1-TMS-vinyl cyclooctanol 2,which was used as obtained for the next step without further purification. Thus,the crude compound 2 was subjected directly to dehydration with POCl3 in pyridine to get the TMS-vinyl substituted cyclooctene 3 in an overall 68% yield. The epoxidation of the cyclodiene 3 proceeded smoothly with a slight surplus of m-CPBA under mild condition in high yield. Epoxide ring-opening and Meinwald rearrangement of epoxide 4 occurred selectively under catalysis of 10 mol% of zinc chloride,leading to the formation of the key intermediate β,γ-unsaturated ketone 5 in a yield of 70%.
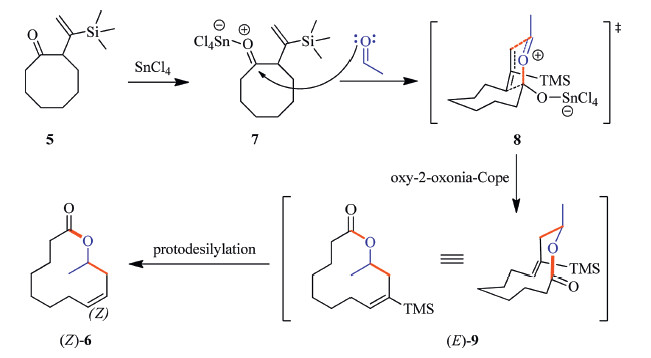
With the 2-TMS-vinylcyclooctanone 5 in hand,we investigated the oxy-2-azonia-Cope reaction applying previously established conditions [5]. To our delight,the rearrangement proceeded smoothly in the presence of an equimolecular stannic tetrachloride, and the desired homoallylic lactone 6 was produced readily in 86% yield. The observed over-all stereochemistry is in accord with the expected mechanism[6TD$DIF] (Scheme 2): [7TD$DIF]The oxy-oxonia-Cope rearrangement produces intermediate (E)-9 via a chair-like transition state 8 in which the oxonium ion occupies an Econfiguration. Although the trimethylsilyl group can survive during the Lewis-acid mediated rearrangement of epoxide 4 to β,γ-unsaturated ketone 5,it is noticeable that the selective protodesilylation of the TMS group occurs after the oxy-2-oxonia Cope rearrangement step. This sharp difference is plausibly due to the presence of water formed from the Lewis acid catalyzed selfcondensation aldol reaction of the excessive acetaldehyde. Related precedents for proto desilylation of vinylsilane moiety in the presence of a Lewis acid have been reported,for example,in the acetal-initiated cyclizations of vinylsilanes to generate allylically unsaturated oxacyclics by Overman [13]. This is advantageous because no additional desylilation had to be performed. It is important to note that the 12-membered ring lactone 6 was obtained exclusively with Z-configuration,maintaining the double bond geometry [11]. The structure of 6 was evidenced by its spectra and HRMS analysis. Since reliable methods [9, 12] for the selective isomerization of (Z)-recifeiolide into (E)-recifeiolide are available,our work offers a new way to this interesting natural product isolated from fungus (Cephalosporium recifei).

|
Download:
|
| Scheme. 2.Mechanism for the oxy-2-azonia-Cope reaction leading to (±)-Z-recifeiolide (6). | |
{kind=link}
In conclusion,we have successfully accomplished the total synthesis of (±)-(Z)-recifeiolide applying a Lewis acid-catalyzed TMS-directed oxy-2-oxonia-Cope rearrangement as the key reaction starting from cyclooctanone. Considering the fact that marcolide skeletons are present in a variety of natural products,the synthetic strategy described herein should provide an efficient way to other natural targets with medium to large sized ring depending on the size of the ring in the starting cycloalkanones. In comparison to more conventional lactonization routes,this formal [n + 4] ring enlargement provides a complementary method to synthesize macrolide target structures. Further studies directed toward related macrolactones employing this protocol are currently carried out in our laboratory.
AcknowledgmentsWe thank G. Tang at Fudan University for the high-resolution MS data. The financial support from the National Natural Science Foundation of China (No. 21372045) is gratefully acknowledged.
| [1] | M. Kerschbaum, Ü ber Lactone mit großen Ringen-die Träger des vegetabilischen Moschus-Duftes, Chem. Ber. 60 (1927) 902-909. |
| [2] | (a) I. Shiina, Total synthesis of natural 8- and 9-membered lactones: recent advancements in medium-sized ring formation, Chem. Rev. 107 (2007) 239-273; (b) G. Rousseau, Medium ring lactones, Tetrahedron 51 (1995) 2777-2849; (c) D. Poth, P.S. Peram, M. Vences, S. Schulz, Macrolides and alcohols as scent gland constituents of the Madagascan frog mantidactylus femoralis and their intraspecific diversity, J. Nat. Prod. 76 (2013) 1548-1558; (d) A. Parenty, X. Moreau, J.M. Campagne, Macrolactonizations in the total synthesis of natural products, Chem. Rev. 106 (2006) 911-939; (e) A. Parenty, X. Moreau, J.M. Gilles Niel, Campagne, Update 1 of: macrolactonizations in the total synthesis of natural products, Chem. Rev. 113 (2013), PR1- PR40; (f) Z.X. Chen, D.L. Liu, W.Y. Gao, T.J. Zhang, A new macrolide and glycosides from the stem of Sargentodoxa cuneate, Chin. Chem. Lett. 20 (2009) 1339-1341. |
| [3] | (a) W. Zhao, Z. Li, J. Sun, A new strategy for efficient synthesis of medium and large ring lactones without high dilution or slow addition, J. Am. Chem. Soc. 135 (2013) 4680-4683; (b) G. Illuminati, L. Mandolini, Ring closure reactions of bifunctional chain molecules, Acc. Chem. Res. 14 (1981) 95-102; (c) A. Gradillas, J. Pé rez-Castells, Macrocyclization by ring-closing metathesis in the total synthesis of natural products: reaction conditions and limitations, Angew. Chem. Int. Ed. 45 (2006) 6086-6101; (d) A. Deiters, S.F. Martin, Synthesis of oxygen- and nitrogen-containing heterocycles by ring-closing metathesis, Chem. Rev. 104 (2004) 2199-2238; (e) C.J. Hou, X.M. Liang, J.P. Wu, D.Q. Wang, Synthesis of macrocyclic lactones with methoxysulfonyl side chain, Chin. Chem. Lett. 19 (2008) 403-405. |
| [4] | D.M. Zubrytski, D.G. Kananovich, O.G. Kulinkovich, A highly stereoselective route to medium-ring-sized trans-alkenolides via oxidative fragmentation of bicyclic oxycyclopropane precursors: application to the synthesis of (+)-recifeiolide, Tetrahedron 70 (2014) 2944-2950. |
| [5] | (a) Y. Zou, H. Mouhib, W. Stahl, et al., Efficient macrocyclization by a novel oxyoxonia- Cope reaction: synthesis and olfactory properties of new macrocyclic musks, Chem. Eur. J. 18 (2012) 7010-7015; (b) Y. Zou, C. Ding, L. Zhou, et al., Tandem cross-dimerisation/oxonia-Cope reaction of carbonyl compounds to homoallylic esters and lactones, Angew. Chem. Int. Ed. 51 (2012) 5647-5651; (c) L. Zhou, Z. Li, Y. Zou, et al., Tandem nucleophilic addition/oxy-2-azonia-Cope rearrangement for the formation of homoallylic amides and lactams: total synthesis and structural verification of motuporamine G, J. Am. Chem. Soc. 134 (2012) 20009-20012; (d) W. Mu, L. Zhou, Y. Zou, Q. Wang, A. Goeke, Irreversible oxy-2-azonia-Cope rearrangements for the synthesis of functionalized allyl a-amino acid derivatives, Eur. J. Org. Chem. (2014) 2379-2385; (e) Y. Zou, L. Zhou, C. Ding, et al., Novel oxy-oxonia(azonia)-Cope reaction: serendipitous discovery and its application to the synthesis of macrocyclic musks, Chem. Biodivers. 11 (2014) 1608-1628. |
| [6] | For isolation and an early synthesis, see: (a) RF. Vesonder, F.H. Stodola, L.J. Wickerham, J.J. Ellis, W.K. Rohwedder, 11- Hydroxy-trans-8-dodecenoic acid lactone, a 12-membered-ring compound from a fungus, Can. J. Chem. 49 (1971) 2029-2032; (b) E.J. Corey, P. Ulrich, J.M. Fitzpatrick, A stereoselective synthesis of (±)-11- hydroxy-trans-8-dodecenoic acid lactone, a naturally occurring macrolide from Cephalosporium recifei, J. Am. Chem. Soc. 98 (1976) 222-224. |
| [7] | X. Huang, C. Craita, L. Awad, P. Vogel, Silyl methallylsulfinates: efficient and powerful agents for the chemoselective silylation of alcohols, polyols, phenols and carboxylic acids, Chem. Commun. 10 (2005) 1297-1299. |
| [8] | H.H. Wasserman, R.J. Gambale, M.J. Pulwer, Activated carboxylates from the photooxygenation of oxazoles: application to the synthesis of recifeiolide, curvularin and macrolides, Tetrahedron 37 (1981) 4059-4067. |
| [9] | J.R. Mahajan, I.S. Resck, Synthesis of (±)-recifeiolide and its homologs via acetylenic lactones, Synth. Commun. 26 (1996) 3809-3819. |
| [10] | A. Fuerstner, K. Langemann, Macrocycles by ring-closing metathesis, Synthesis (1997) 792-803. |
| [11] | K. Okuma, S. Hirabayashi, M. Ono, K. Shioji, H. Matsuyama, H.J. Bestmann, An efficient synthesis of (R)-(+)-recifeiolide and related macrolides by using enantiomerically pure (R)-(-)-5-methyl-2,2,2-triphenyl-1,2λ5-oxaphospholane, Tetrahedron 54 (1998) 4243-4250. |
| [12] | I. Fleming, A. Barbero, D. Walter, Stereochemical control in organic synthesis using silicon-containing compounds, Chem. Rev. 97 (1997) 2063-2192. |
| [13] | L.E. Overman, A. Castaneda, T.A. Blumenkopf, Acetal-initiated cyclizations of vinylsilanes. A general synthesis of allylically unsaturated oxacyclic, J. Am. Chem. Soc. 108 (1986) 1303-1304. |