 2015, Vol.26
2015, Vol.26 



b Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 201814, China
The γ-butyrolactone is a characteristic skeletal core present in a large amount of natural products and biological compounds including pharmaceuticals and agrochemicals,such as ascorbic acid [1],sex pheromones [2],plant growth regulator karrikin [3] and anti-HIV nucleoside β-FddA [4]. Development of efficient methods for the synthesis of various functionalized lactones has attracted extensive attention [5, 6, 7, 8, 9].
As we know,the introduction of fluorine-containing groups into organic molecules could alter their properties and bioactivities [10, 11, 12, 13, 14]. The synthesis of fluorine-containing γ-butyrolactones is of current interests [15, 16, 17, 18, 19]. Despite this interest,general synthetic methods for the incorporation of the gem-difluoromethylene group into γ-butyrolactones are still highly desirable [20, 21]. Pohmakotr et al. have disclosed the synthesis of gem-difluoromethylenated spiro-γ-butyrolactones by using PhSCF2Si(CH3)3 as a gem-difluoromethylenating building block [20]. 4,5-Allenoic acids reacted with selectfluor to provide the desired 1,1-difluoroallyl γ-butyrolactones was reported by Zhao’s group [21].
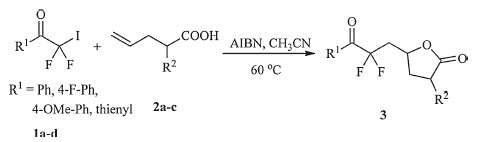
In recent years,our laboratory has been devoted to prepare polyfluoroalkyl-containing lactones from the free radical additions of polyfluoroalkyl iodides and 5-hexenoic acids/4-pentenoic acid initiated by Na2S2O4 [22, 23] and Pd(PPh3)4 [24]. As part of our continued interest in expanding the structural diversity of the fluoroalkyl-containing lactones,herein,we reported our new results regarding the synthesis of difluoroalkyl-containing γ-butyrolactones via the free radical addition of iododifluoromethyl ketones to 4-pentenoic acids initiated by 2,2-azobisisobutyronitrile (AIBN) in CH3CN at 60 ℃ (Scheme 1).

|
Download:
|
| Scheme. 1.Synthesis of difluoroalkyl-containing g-butyrolactones. | |
All reagents were of analytical grade,and obtained from commercial suppliers and used without further purification. All NMR spectra were recorded on a Bruker Avance 500 (resonance frequencies 500 MHz for 1H and 126 MHz for 13C) equipped with a 5 mm inverse broadband probe head with z-gradients at 295.8 K with standard Bruker pulse programs. The samples were dissolved in 0.6 mL CDCl3 (99.8% D.TMS). Chemical shifts were given in values of δH and δC referenced to residual solvent signals (δH 7.26 for 1H,δC 77.0 for 13C in CDCl3). The 19F NMR spectra were obtained using a 500 spectrometer (471 MHz) using trifluorotoluene as external standard. High resolution mass spectra (HRMS) were recorded on a Bruker solan X 70 FT-MS (samples was dissolved in CH3OH and the ion source was ESI),and the energy was 22.5 eV at MS/MS. Melting points are uncorrected.
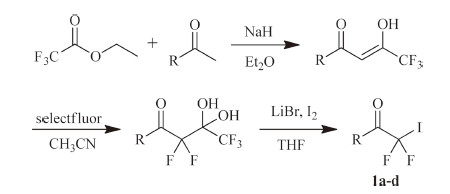
2.1. Preparation of iododifluoromethyl ketones 1a-dThe iododifluoromethyl ketones 1a-d were prepared according to the reported procedure [25, 26, 27]. As shown in Scheme 2,the intermediates enols were obtained from the reaction of trifluoromethyl ethyl acetate and ketones. The enols reacted with Selectfluor® to form fluorinated gem-diols,which then reacted with I2 to afford iododifluoromethyl ketones 1a-d using the trifluoroacetate release conditions (Scheme 2).

|
Download:
|
| Scheme. 2.Synthesis of iododifluoromethyl ketones 1a–d. | |
To a solution of iododifluoromethyl ketones 1a-d (1.5 mmol) in acetonitrile (10 mL) was added 4-pentenoic acids (1 mmol) and AIBN (33 mg,20 mmol%) at room temperature, the mixture was then heated to 60 ℃ and stirred for 14-16 h (TLC). After the completion of reaction,the reaction was quenched with H2O. The mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with brine,dried over anhydrous Na2SO4,and concentrated under reduced pressure. The residue was purified by flash column chromatography using petroleum ether/EtOAc (10:1) as eluent to afford the corresponding difluoroalkyl-containing lactones 3.
5-(2,2-Difluoro-3-oxo-3-phenylpropyl)dihydrofuran-2(3H)- one (3aa): White solid,yield: 82%,mp: 49.5-50.7 ℃. 1H NMR (500 MHz,CDCl3): δ 8.13-7.51 (m,5H),4.90-4.86 (m,1H),2.85- 2.74 (m,1H),2.62-2.48 (m,4H),2.10-2.01(m,1H); 13C NMR (126 MHz,CDCl3): δ 188.5 (t,2JC-F = 30.8 Hz),176.1,134.6,131.5, 130.2 (t,3JC-F = 3.0 Hz),128.8,118.2 (t,1JC-F = 255.1 Hz),74.5 (t, 3‘JC-F = 4.2 Hz),39.3 (t,2’JC-F = 22.4 Hz),28.8,28.4; 19F NMR (471 MHz,CDCl3): δ -98.4 (AB,2JF-F = 296.3 Hz,2F). HRMS calcd. for C13H12F2O3:254.0755,found: 254.0762.
5-(2,2-Difluoro-3-oxo-3-phenylpropyl)3,3-dimethyldihydrofuran- 2(3H)-one (3ab): White solid,yield: 70%,mp: 57.3-58.1 ℃. 1H NMR (500 MHz,CDCl3): δ 8.13-7.51(m,5H),4.84-4.79 (m,1H), 2.84-2.72 (m,1H),2.58-2.51(m,1H),2.37-2.33 (m,1H),1.94-1.90 (m,1H),1.31-1.29 (m,6H); 13C NMR (126 MHz,CDCl3): δ 188.5 (t,2JC-F = 30.7 Hz),181.0,134.6,131.4,130.2,128.8,118.2 (t, 1JC-F = 254.9 Hz),70.8 (t,3’JC-F = 4.3 Hz),44.1,40.0,39.5 (t, 20JC-F = 22.7 Hz),24.9,24.1; 19F NMR (471 MHz,CDCl3): δ -98.4 (AB,2JF-F = 296.6 Hz,2F). HRMS calcd. for C15H16F2O3:282.1068, found: 282.0958.
5-(2,2-Difluoro-3-(4-fluorophenyl)-3-oxopropyl)dihydrofuran- 2(3H)-one (3ba): Yellow oil liquid,yield: 73%. 1H NMR (500 MHz,CDCl3): δ 8.17-7.17 (m,4H),4.90-4.86 (m,1H),2.83- 2.71 (m,1H),2.62-2.50 (m,4H),2.10-2.01(m,1H); 13C NMR (126 MHz,CDCl3): δ 186.9 (t,20JC-F = 31.3 Hz),176.1,166.6 (δ, 1JC-F = 258.1 Hz),133.2 (dt,4JC-F = 9.4 Hz,3'JC-F = 3.1 Hz),127.8, 118.4 (t,1'JC-F = 255.1 Hz),116.1 (δ,2JC-F = 22.1 Hz),74.4 (t, 3''JC-F = 4.2 Hz),39.1(t,2''JC-F = 22.4 Hz),28.8,28.4; 19F NMR (471 MHz,CDCl3): δ -98.2 (AB,2JF-F = 298.4 Hz,2F),-101.6 (s, 1F). HRMS calcd. for C13H11F3O3: 272.0660,found: 272.0647.
5-(2,2-Difluoro-3-(4-fluorophenyl)-3-oxopropyl)-3,3- dimethyldihydrofuran-2(3H)-one (3bb): Yellow oil liquid,yield: 65%. 1H NMR (500 MHz,CDCl3): δ 8.18-4.78(m,4H),4.83-4.78 (m, 1H),2.81-2.51 (m,2H),2.36-1.89 (m,2H),1.33-1.29 (m,6H); 13C NMR (126 MHz,CDCl3): δ 186.9 (t,20JC-F = 31.1 Hz),181.0,166.6 (d, 1JC-F = 258.3 Hz),133.2 (dt,4JC-F = 9.7 Hz,3’JC-F = 3.0 Hz),127.8, 118.2 (t,1‘JC-F = 255.0 Hz),116.1 (d,2JC-F = 22.0 Hz),70.8 (t, 3’’JC-F = 3.8 Hz),44.1,40.0,39.4 (t,2’’JC-F = 22.4 Hz),24.8,24.0; 19F NMR (471 MHz,CDCl3): δ -98.1(AB,2JF-F = 298.4 Hz,2F), -101.6 (s,1F). HRMS calcd. for C15H15F3O3: 300.0973,found: 300.0939.
5-(2,2-Difluoro-3-(4-methoxyphenyl)-3-oxopropyl)dihydrofuran- 2(3H)-one (3ca): White solid,yield: 84%,mp: 80.8-81.4 ℃. 1H NMR (500 MHz,CDCl3): δ 8.12-6.98 (m,4H),4.90-4.83(m,1H), 3.91(s,3H),2.79-2.72 (m,1H),2.58-2.50 (m,4H),2.08-2.04 (m, 1H); 13C NMR (126 MHz,CDCl3): δ 186.8 (t,2JC-F = 30.6 Hz),176.3, 164.8,124.2,115.4 (t,1JC-F = 254.9 Hz),114.1,74.7 (t, 3'JC-F = 3.8 Hz),55.6,39.4 (t,2'JC-F = 22.2 Hz),28.9,28.5; 19F NMR (471 MHz,CDCl3): δ -96.9 to -98.8 (m,2F). HRMS calcd. for C14H14F2O4: 284.0860,found: 284.0932.
5-(2,2-Difluoro-3-(4-methoxyphenyl)-3-oxopropyl)-3,3- dimethyldihydrofuran-2(3H)-one (3cb): White solid,yield: 71%, mp: 81.4-82.1 ℃. 1H NMR (500 MHz,CDCl3): δ 8.10-6.95 (m,4H), 4.81-4.76 (m,1H),3.88 (s,3H),2.78-2.47 (m,2H),2.33-1.86 (m, 2H),1.27-1.26 (m,6H); 13C NMR (126 MHz,CDCl3): δ 186.8 (t, 2JC-F = 30.4 Hz),181.1,164.8,132.8 (t,3JC-F = 3.0 Hz),124.2,118.4 (t,1JC-F = 254.9 Hz),114.1,80.0 (t,3'JC-F = 4.2 Hz),55.6,44.2,40.0, 39.6 (t,2'JC-F = 22.2 Hz),24.9,24.1; 19F NMR (471 MHz,CDCl3): δ -97.7 to -97.9 (m,2F). HRMS calcd. for C16H18F2O4: 312.1173, found: 312.1245.
5-(2,2-Difluoro-3-(4-methoxyphenyl)-3-oxopropyl)-3-methyldihydrofuran- 2(3H)-one (3cc): White solid,yield: 79%,mp: 81.7- 82.1 ℃. 1H NMR (500 MHz,CDCl3): δ 8.12-6.97(m,4H),4.92-4.70 (m,0.5H + 0.5H),3.90 (s,3H),2.78-2.47 (m,4H),2.32-2.13 (m,1H), 1.31 (d,1.5H,J = 7.9 Hz),1.29 (d,1.5H,J = 7.5 Hz); 13C NMR (126 MHz,CDCl3): δ 186.8 (t,2JC-F = 30.4 Hz),179.1 (s,0.5C),178.6 (s,0.5C),164.8,132.8,124.2,118.4 (t,1JC-F = 254.9 Hz),114.1,72.4 (t,3’JC-F = 4.2 Hz,0.5C),72.3 (t,30JC-F = 4.2 Hz,0.5C),55.6,39.4 (t, 20JC-F = 22.6 Hz,0.5C),39.1 (t,20JC-F = 23.0 Hz,0.5C),38.0 (s,0.5C), 36.1 (s,0.5C),35.5 (s,0.5C),33.7 (s,0.5C),15.6 (s,0.5C),14.9 (s, 0.5C); 19F NMR (471 MHz,CDCl3): δ -96.8 to -99.1 (m,1F),97.8 (s, 1F). HRMS calcd. for C15H16F2O4: 298.1017,found: 298.0905.
5-(2,2-Difluoro-3-oxo-3-(thiophen-2-yl)propyl)dihydrofuran- 2(3H)-one (3da): White solid,yield: 80%,mp: 71.6-72.3 ℃. 1H NMR (500 MHz,CDCl3): δ 8.04-7.21 (m,3H),4.86-4.83 (m,1H), 2.80-2.69 (m,1H),2.60-2.48 (m,4H),2.08-1.99 (m,1H); 13C NMR (126 MHz,CDCl3): δ 181.8 (t,2JC-F = 30.4 Hz),171.6,137.5,137.0, 136.2 (t,3JC-F = 4.6 Hz),129.0,117.7 (t,1JC-F = 254.6 Hz),74.4 (t, 3'JC-F = 3.8 Hz),39.3 (t,20JC-F = 22.5 Hz),28.8,28.4; 19F NMR (471 MHz,CDCl3): δ -99.4 (AB,2JF-F = 284.7 Hz,2F). HRMS calcd. for C11H10F2O3S:260.0318,found: 260.0391.
5-(2,2-Difluoro-3-oxo-3-(thiophen-2-yl)propyl)-3,3-dimethyldihydrofuran- 2(3H)-one (3db): White solid,yield: 71%,mp: 95.9- 97.0 ℃. 1H NMR (500 MHz,CDCl3): d 8.05-7.22 (m,3H),4.82-4.76 (m,1H),2.79-2.68 (m,1H),2.57-2.46 (m,1H),2.35-2.32 (m,1H), 1.93-1.88 (m,1H),1.30-1.28 (m,6H); 13C NMR (126MHz,CDCl3): d 181.9 (t,2JC-F = 30.4 Hz),181.0,137.5,137.0,136.2 (t,3JC-F = 5.1 Hz), 128.9,117.7 (t,1JC-F = 254.6 Hz),70.4 (t,30JC-F = 4.2 Hz),44.0,40.0, 39.5 (t,20JC-F = 22.5 Hz),24.8,24.1; 19F NMR (471MHz,CDCl3): d -99.3 (AB,2JF-F = 289.4 Hz,2F). HRMS calcd. for C13H14F2O3S: 288.0632,found: 288.0708.
5-(2,2-Difluoro-3-oxo-3-(thiophen-2-yl)propyl)-3-methyldihydrofuran- 2(3H)-one (3dc): White solid,yield: 66%,mp: 54.4- 55.0 8C. 1H NMR (500 MHz,CDCl3): δ 8.04-7.21(m,3H),4.92-4.69 (m,0.5H + 0.5H),2.80-2.45 (m,4H),2.31-2.14 (m,1H),1.30 (d,1.5H, J = 7.9 Hz),1.29 (d,1.5H,J = 7.0 Hz); 13C NMR (126MHz,CDCl3): δ 181.9 (t,2JC-F = 31.5 Hz),179.0 (s,0.5C),178.6 (s,0.5C),137.5,137.0, 136.2 (t,3JC-F = 4.6 Hz),129.0,117.7 (t,1JC-F = 254.6 Hz,0.5C),117.6 (t,1JC-F = 254.6 Hz,0.5C),72.1 (t,3'JC-F = 4.0 Hz,0.5C),72.0 (t, 3'JC-F = 4.3 Hz,0.5C),39.4 (t,2'JC-F = 22.6 Hz),37.9 (s,0.5C),36.0 (s, 0.5C),35.5 (s,0.5C),33.6 (s,0.5C),15.6 (s,0.5C),14.8 (s,0.5C); 19F NMR (471MHz,CDCl3): δ -98.5 to -100.0 (m,1F),-98.7 to -100.4 (m,1F); HRMS calcd. for C12H12F2O3S: 274.0475,found: 274.0544.
6-(2,2-Difluoro-3-oxo-3-phenylpropyl)tetrahydro-2H-pyran- 2-one (3ad): Light white oil,yield: 77%. 1H NMR (500 MHz,CDCl3): δ 8.12-7.51 (m,5H),4.41-4.36 (m,1H),3.17-3.05 (m,1H),3.00- 2.89 (m,1H),2.46-2.42 (m,2H),1.95-1.87 (m,3H),1.81-1.78 (m, 1H); 13C NMR (126 MHz,CDCl3): δ 188.5 (t,2JC-F = 30.9 Hz),179.0, 134.6,131.6,130.2,128.8,118.8 (t,1JC-F = 256.2 Hz),44.5 (t, 2'JC-F = 22.2 Hz),39.7,32.9,24.8,22.1; 19FNMR(471 MHz,CDCl3): δ -98.80 (AB,2JF-F = 291.6 Hz,2F). HRMS calcd. for C14H14F2O3: 268.0911,found: 268.0803.
6-(2,2-Difluoro-3-(4-fluorophenyl)-3-oxopropyl)tetrahydro-2Hpyran- 2-one (3bd): Yellow oil liquid,yield: 69%. 1H NMR (500MHz,CDCl3): δ 8.13-7.15 (m,4H),4.38-4.32 (m,1H),3.08- 3.05 (m,1H),2.93-2.86 (m,1H),2.43-2.42 (m,2H),1.89-1.73 (m, 4H); 13C NMR (126 MHz,CDCl3): δ 186.9 (t,2JC-F = 30.5 Hz),179.4, 166.5 (d,1'JC-F = 258.2 Hz),133.2 (d,3'JC-F = 9.0 Hz),127.9,118.8 (t, 1JC-F = 256.1 Hz),116.1 (t,20JC-F = 22.1 Hz),44.2(t,200JC-F = 21.9 Hz), 39.7,32.9,24.8,22.0; 19FNMR (471 MHz,CDCl3): δ-97.38 to-99.28 (m,2F),-99.6 (s,1F). HRMS calcd. for C14H13F3O4: 286.0817,found: 286.0832.
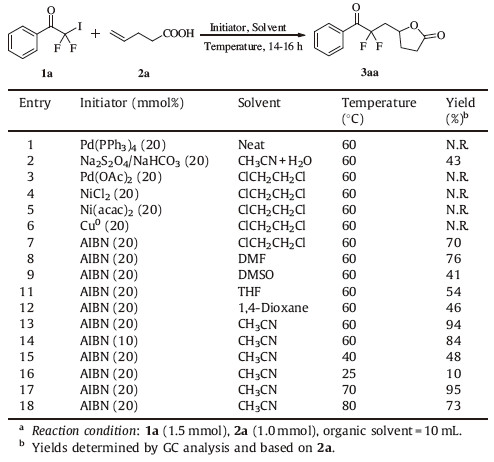
3. Results and discussionOn the basis of previous literature,introduction of difluoromethylene group into organics could be accomplished by the radical addition of iododifluoromethyl ketones to alkenes initiated by Pd(PPh3)4 [28],Cu catalyst [29] and UV irradiation [30]. Therefore,we tried to use iododifluoromethyl ketones as the difluoromethyl radicals in this reaction. The iododifluoromethyl ketones in this communication were prepared by using release/ halogenation protocol reported by Colby [27]. Initially,we carried out the reaction of 2,2-difluoro-2-iodo-1-phenylethanone 1a with 4-pentenoic acid 2a in the presence of Pd(PPh3)4 at room temperature under solvent-free condition [28]. However,the reaction hardly proceeded and a large amount (70%) of the starting materialwasrecovered along with the formation of the side product 2,2-difluoro-1-phenylethanone without any desired product 3a (Table 1,entry 1). The result might be ascribed to the substrate specificity of iododifluoromethyl ketone. When Na2S2O4/NaHCO3 were used as initiators,the product difluoroalkyl-g-butyrolactone 3a was obtained in moderate yield under the reported reaction condition [22] by using CH3CN/H2O as solvent at 60 ℃ (entry 2), whereas Pd,Ni or Cu catalyst still afford no desire product in ClCH2CH2Cl (entries 3-6). As AIBN provided a relatively higher yield of 3a than Na2S2O4/NaHCO3 (entry 7 vs. entry 2),we chose AIBN as the initiator for further optimization of reaction conditions.
| Table 1 Optimization of reaction conditions for the synthesis of compound 3aa.a |
{kind=link}
{kind=link}
Next,the influence of solvents on the model reaction was investigated (entries 8-13). CH3CN promoted the reaction much more efficiently and gave a high yield of the product 3aa (entry 13). Decreasing the amount of AIBN to 10 mmol% led to a decrease in yield of 3aa (entry 14). In addition,the raising of the reaction temperature did not promoted the reaction (entries 15 and 16). The reaction proceeded less efficiently when the reaction temperature was decreasing (entries 17 and 18).
Having established the optimized method,we next probe the generality and scope of this radical addition reaction. Several iododifluoromethyl ketones and different 4-pentenoic acids were examined under the optimized reaction conditions (Scheme 3). In most cases,the radical addition reaction proceeded smoothly to furnish the difluoroalkyl-containing γ-butyrolactones in good yields,and no primary adduct β-iodo(perfluoroalkyl)-alkanoic acid was observed. When 2-methylpent-4-enoic acid was used as substrate to react with 2,2-difluoro-2-iodo-1-(4-methoxyphenyl) ethanone,a mixture of trans- and cis-isomers of the corresponding difluoroalkyl-containing γ-butyrolactone was obtained in almost a 1:1 ratio (determined by 1H NMR),and the two isomers could not be separated due to their similarity in affinity to chromatographic silica gel.

|
Download:
|
| Scheme. 3.Reactions scope of iododifluoromethyl ketones and 4-pentenoic acids. | |
{kind=link}
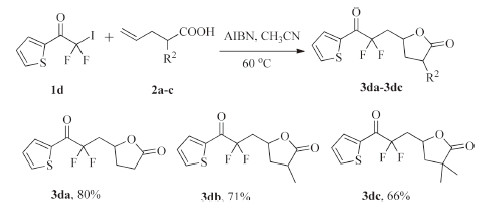
Next,the radical addition reactions of 2,2-difluoro-2-iodo- 1-(thiophen-2-yl) ethanone with 4-pentenoic acid and 2,2- dimethylpent-4-enoic acid gave the difluoroalkyl-containing g-butyrolactones in good yields. 2,2-Difluoro-2-iodo-1-(thiophen- 2-yl)ethanone reactedwith 2-methylpent-4-enoic acid also provided two isomers of 5-(2,2-difluoro-3-oxo-3-(thiophen-2- yl)propyl)-3,3-dimethyldihydro furan-2(3H)-one (Scheme 4).

|
Download:
|
| Scheme. 4.Reactions scope of 2,2-difluoro-2-iodo-1-(thiophen-2-yl)ethanone and 4-pentenoic acids. | |
{kind=link}
Finally,difluoroalkyl-containing δ-lactones were synthesized efficiently from the radical addition reactions of 2,2-difluoro-2- iodo-1-phenylethanone derivatives and 5-hexenoic acid under the mentioned reaction condition (Scheme 5).

|
Download:
|
| Scheme. 5.Reactions scope of 2,2-difluoro-2-iodo-1-phenylethanone derivatives and 5-hexenoic acid. | |
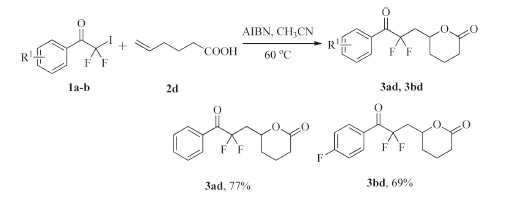{kind=link}
Considering that aliphatic iododifluoromethyl ketones were inconveniently prepared by the protocol used in this paper,we reported herein the radical addition reactions of aromatic iododifluoromethyl ketones and 4-pentenoic acids/5-hexenoic acid. The reactions with aliphatic iododifluoromethyl ketones are under way.
4. ConclusionIn summary,a convenient and efficient approach for difluoroalkyl- containing g-butyrolactones via the radical addition reaction of iododifluoromethyl ketones with 4-pentenoic acids initiated by AIBN has been developed through sequential radical difluoroalkylation and nucleophilic cyclization. The difluoroalkylcontaining d-valerolactones were also synthesized efficiently under this reaction conditions.
AcknowledgmentWe are grateful for financial supports from the National Natural Science Foundation of China (Nos. 21472126,21172148, 21302128).
| [1] | R.P. Tripathi, B. Singh, S.S. Bisht, J. Pnadey, L-Ascorbic acid in organic synthesis: an overview, Curr. Org. Chem. 13 (2009) 99-122. |
| [2] | C. Wang, G.A. Russell, γ-Lactone formation in the addition of benzenesulfonyl bromide to diene and enyne esters, J. Org. Chem. 64 (1999) 2066-2069. |
| [3] | G.R. Flematti, E.L. Ghisalberti, K.W. Dixon, R.D. Trengove, A compound from smoke that promotes seed germination, Science 305 (2004),977. |
| [4] | C. Meier, T. Knispel, V.E. Marquez, et al., Cyclosal-pro-nucleotides of 2'-fluoro-araand 2'-fluoro-ribo-2',3'-dideoxyadenosine (F-ara- and F-ribo-ddA) as a strategy to bypass a metabolic blockade, J. Med. Chem. 42 (1999) 1615-1624. |
| [5] | M.A. Zeller, M. Riener, D.A. Nicewicz, Butyrolactone synthesis via polar radical crossover cycloaddition reactions: diastereoselective syntheses of methylenolactocin and protolichesterinic acid, Org. Lett. 18 (2014) 4810-4813. |
| [6] | T.P. Montgomery, A.H. Boyoung, Y. Park, M.J. Krische, Enantioselective conversion of primary alcohols to α-exo-Methylene γ-butyrolactones via iridium-catalyzed C-C bond-forming transfer hydrogenation: 2-(alkoxycarbonyl)allylation, J. Am. Chem. Soc. 27 (2012) 11100-11103. |
| [7] | S. Li, S. Ma, Highly selective nickel-catalyzed methyl-carboxylation of homopropargylic alcohols for α-alkylidene-γ-butyrolactones, Org. Lett. 22 (2011) 6046- 6049. |
| [8] | T.M. Lv, F.H. Wu, Progress in selective iodolactonization, Chin. J. Org. Chem. 8 (2003) 763-769. |
| [9] | M. Zhu, L. Li, J.Y. Tang, H. Zhang, An effective method for the preparation of chlorolactones, Chin. Chem. Lett. 4 (2011) 431-434. |
| [10] | B.E. Smart, Fluorine substituent effects on bioactivity, J. Fluorine Chem. 109 (2007) 3-11. |
| [11] | S. Purser, P.R. Moore, S. Swallow, V. Gouverneur, Fluorine in medicinal chemistry, Chem. Soc. Rev. 37 (2008) 320-330. |
| [12] | X.Y. Jiang, X.H. Xu, F.L. Qing, Design and concise synthesis of gem-difluormethylenated analogue of 7-epi-castanospermine, Chin. Chem. Lett. 8 (2014) 1115- 1120. |
| [13] | W. Gu, J. Lin, J. Xiao, Direct N-gem-difluorocyclopropylation of nitro-heterocycles by utilizing gem-difluorocyclopropyl tosylate, Chin. Chem. Lett. 24 (2014) 24-28. |
| [14] | C. Lu, Y.L. Shi, Z. Zhou, et al., Perfluorinated compounds in blood of textile workers and barbers, Chin. Chem. Lett. 25 (2014) 1145-1148. |
| [15] | H. Xiao, H.Y. Duan, R.S. Yao, et al., Chemoselective synthesis of trifluoromethylated γ-butenolide derivatives via phosphine-promoted tandem reaction of allylic carbonates and trifluoromethyl ketones, Org. Lett. 20 (2014) 5462-5465. |
| [16] | Y. Xiong, Z.H. Ju, X.G. Wang, X. Fang, F.H. Wu, Progress in the synthetic methods of fluorine-containing lactones, Chin. J. Org. Chem. 11 (2009) 1728-1743. |
| [17] | P. Bravo, M. Frigerio, A. Melloni, et al., Stereoselective synthesis of both enantiomers of trifluoro-γ-valerolactone and pentafluoro-γ-caprolactone, Eur. J. Org. Chem. (2002) 1895-1902. |
| [18] | K. Tenza, J.S. Northen, D. O'Hangan, A.M.Z. Slawin, Stereoselective α-fluoroamide and a-fluoro-γ-lactone synthesis by an asymmetric zwitterionic aza-claisen rearrangement, Beilstein J. Org. Chem. 13 (2005) 13-17. |
| [19] | M. Tredwell, J.A.R. Luft, M. Schuler, et al., Fluorine-directed diastereoselective iodocyclizations, Angew. Chem. Int. Ed. 2 (2008) 357-360. |
| [20] | A. Chatupheeraphat, D. Soorukram, C. Kuhakarn, et al., Synthesis of gem-difluoromethylenated spiro-γ-butyrolactones by employing PhSCF2Si(CH3)3 as a gemdifluoromethylenating agent, Eur. J. Org. Chem. (2013) 6844-6858. |
| [21] | H.F. Cui, Z. Chai, Y.P. Lu, et al., Direct double electrophilic fluorination of allenoic acids and tosylamides to give 1,1-difluoroallylic heterocyclic compounds, Chin. J. Chem. 29 (2011) 2744-2748. |
| [22] | X. Fang, Q. Ying, Y. Chen, et al., Synthesis of fluoroalkyl-δ-lactones from polyfluoroalkyl iodides and 5-hexenoic acids, J. Fluorine Chem. 128 (2008) 280-285. |
| [23] | X.W. Zou, F.H. Wu, A convenient method of polyfluoroalkyl-lactonization, Chin.Chem. Lett. 13 (2002) 410-411. |
| [24] | F.H. Xiao, F.H. Wu, X.Y. Yang, Y.J. Shen, X.M. Shi, A convenient synthesis of fluoroalkylated γ-butyrolactones from polyfluoroalkyl iodides and 4-pentenoic acid catalyzed by Pd(PPh3)4, J. Fluorine Chem. 126 (2005) 319-323. |
| [25] | I. Saidalimu, X. Fang, X.P. He, et al., Highly hnantioselective construction of 3-hydroxy oxindoles through a decarboxylative aldol addition of trifluoromethyl α-fluorinatedgem-diols to N-benzyl isatins, Angew. Chem. Int. Ed. 52 (2013) 5566-5570. |
| [26] | P. Zhang, C. Wolf, Synthesis of pentafluorinated β-hydroxy ketones, J. Org. Chem. 79 (2012) 8840-8844. |
| [27] | J.P. John, D.A. Colby, Synthesis of α-halo-α,α-difluoromethyl ketones by a trifluoroacetate release/halogenation protocol, J. Org. Chem. 78 (2011) 9163-9168. |
| [28] | Z.M. Qiu, D.J. Burton, A general route to a,a-difluoroketones, Tetrahedron Lett. 34 (1993) 3239-3242. |
| [29] | K.C. Kwak, H. Oh, Y.G. Yun, et al., Addition of a,a-difluoroiodomethylcyclohexyl ketone to alkenes under copper catalyst, Bull. Korean Chem. 23 (2002) 157-159. |
| [30] | Z.M. Qiu, D.J. Burton, Synthesis of γ-(electron-withdrawing group)-substituted aa-difluoro ketones by UV-initiated addition of iododifluoromethyl ketones with electron-deficient alkenes, J. Org. Chem. 60 (1995) 6798-6805. |