 2015, Vol.26
2015, Vol.26 



b State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing 210009, China
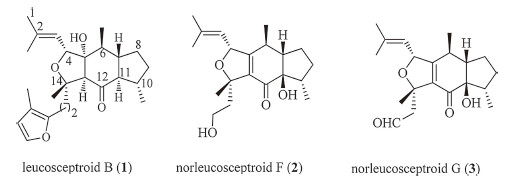
Since 2010,a family of terpenoids,named as leucosceptroids and norleucosceptroids,have been isolated and identified from Leucosceptrum canyn by Li’s group [1]. Additionally,several structurally similar sesterterpenoids were isolated from Colquhounia coccinea by the same group [2]. This family of terpenoids shows intriguing antifeedant and antifungal activity. As shown by representative molecular structures in Fig. 1,these terpenoids embrace a typical 5/6/5 tricyclic core framework decorated with stereogenic centers. Different side chains are attached to the fully functionalized dihydrofuran or tetrahydrofuran. The complex molecular architecture of these hydrindane-containing terpenoids [3],as well as their interesting bioactivity,make them challenging synthetic targets for chemists [4].

|
Download:
|
| Fig. 1.Molecular structures of leucosceptroid B, norleucosceptroids F and G. | |
{kind=link}
Horne’s group reported their model construction of the tricyclic framework of leucosceptroid family by employing intramolecular Diels-Alder cycloaddition as the key step [4a]. In 2012,our group accomplished the first total synthesis of the member among this terpenoid family,leucosceptroid B,by developing an oxa-Michael cyclization-dehydration-deprotection cascade as the key step to install the tricyclic core [4b]. Recently,Magauer’s group employed a formal intermolecular [4 + 2] cycloaddition and an unprecedented dilactol aldol-type condensation to complete the total synthesis of norleucosceptroids A-B and leucosceptroid K [4c],and collective total synthesis of other members in this family [4d, 4e]. By adopting a convergent strategy featured with aldol reaction and SmI2-mediated ketyl-olefin cyclization,Ma’s group completed total synthesis of leucosceptroids A-B [4f]. Herein,we present our effort on total synthesis of norleucosceptroids F and G (Fig. 1) (Scheme 1).
|
|
Download:
|
| Scheme 1.The synthesis of norleucosceptroid F (2) and norleucosceptroid G (3). | |
All reactions were carried out under an argon atmosphere with dry solvents under anhydrous conditions,unless otherwise noted. Tetrahydrofuran was distilled from sodium-benzophenone and dichloromethane was distilled from calcium hydride. All the chemicals were purchased commercially and used without further purification,unless otherwise stated. Flash chromatography was performed using silica gel (200-400 mesh). Reactions were monitored by thin layer chromatography (TLC). Visualization was achieved under a UV lamp (254 nm and 365 nm),I2 and by developing the plates with p-anisaldehyde or phosphomolybdic acid. 1H NMR and 13C NMR were recorded on Bruker DRX-400 MHz NMR,Bruker DRX-600 MHz NMR spectrometer with TMS as the internal standard and were calibrated using residual undeuterated solvent as an internal reference. High resolution mass spectra (HRMS) were recorded by using FTMS-7 spectrometers.
Preparation of (6): To a solution of 5 (1.22 g,6.6 mmol) in THF (75 mL) was added n-BuLi (2.4 mol/L in hexane,3.1 mL,7.3 mmol) at -78℃,the mixture was stirred for 1 h. A solution of 4 (1.0 g,6.0 mmol) in THF (6 mL) was added in one portion and the mixture was stirred for 10 min at -78℃ then quenched with saturated aqueous NH4Cl (50 mL). The layers were separated and the aqueous phase was extracted with EtOAc (2 × 60 mL). The organic layer was dried over Na2SO4,filtered,and concentrated to afford yellow oil as the crude product. To the mixture of this yellow oil in DCM (60 mL) was added TEMPO (46.8 mg,0.3 mmol) and PhI(OAc)2 (3.9 g,12.0 mmol) at 0℃. The reaction was stirred at r.t. overnight then quenched with saturated aqueous Na2SO3 (50 mL) and saturated aqueous NaHCO3 (50 mL). The layers were separated and the aqueous phase was extracted with CH2Cl2 (2 × 70 mL),the combined organic layers were dried over MgSO4,filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (10:1,PE/ EtOAc) to provide 6 (1.13 g,53.3% over two steps) as a pale yellow oil. [a]D 26 15.4 (c 0.25,CHCl3); IR (thin film,cm-1): 3145,2959,1724,1658,1393,1257,1105; 1HNMR(400 MHz,CDCl3): δ 9.69 (d,1H,J = 1.6 Hz),3.76 (t,2H,J = 6.8 Hz),3.39 (t,1H,J = 6.4 Hz),2.56 (t,2H,J = 6.8 Hz),2.33-2.24 (m,1H),2.15-2.05 (m,1H),1.94-1.70 (m,3H),1.57-1.48 (m,1H),1.33-1.27 (m,1H),1.08 (d,3H,J = 2.0 Hz),1.06 (d,3H,J = 2.0 Hz),0.89 (s,9H),0.07 (s,6H); 13CNMR(100 MHz,CDCl3): δ 204.4,190.9,91.5,84.1,60.9,60.0,48.1,45.6,40.1,30.6,28.8,26.0,23.6,18.4,16.4,13.5,-5.2; HRMS (ES) m/z calcd. for C20H34O3SiNa (M + Na)+ 373.2175,found 373.2172.
Preparation of (7): To a stirred solution of methoxymethyl allyl ether (545.0 mg,5.3 mmol) in THF (20 mL) was added s-BuLi (1.3 mol/L in hexane,4.0 mL,5.3 mmol) at -78℃ slowly. After the resultant orange yellow solution was stirred at -78℃ for 30 min,(-)-B-methoxydiisopinocamphenylborane (2.0 mol/L in THF,2.3 mL,4.6 mmol) was added. After the addition was completed,the mixture was stirred at -78℃ for 1 h and BF3.Et2O (0.6 mL,4.7 mmol) was added dropwise. Immediately afterwards,a solution of 6 (910.0 mg,2.6 mmol) in THF (10 mL) was added dropwise,then the mixture was warmed to -50℃ slowly and the mixture was warmed to r.t. immediately,then saturated aqueous NaHCO3 (40 mL) was added followed by 30% aqueous H2O2 (30 mL),and the resulting mixture was stirred for 10 h at r.t. The aqueous layer was extracted with EtOAc (2 × 100 mL) and the combined organic layers were then dried over Na2SO4,filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (10: 1,PE/EtOAc) to provide 7 (544 mg,46.3%) as a pale yellow oil. [a]D 30 -62.1 (c 0.31,CHCl3); IR (thin film,cm-1): 3445,2955,2932,2885,2210,1662,1384,1256,1105,1030,919,833,779,669; 1H NMR (400 MHz,CDCl3): δ 5.49 (ddd,1H,J = 16.4,10.8,8.4 Hz),5.30 (s,1H),5.27 (dd,1H,J = 8.4,1.2 Hz),4.73 (d,1H,J = 6.8 Hz),4.56 (d,1H,J = 6.8 Hz),3.90 (t,1H,J = 8.8 Hz),3.76 (t,2H,J = 6.8 Hz),3.64 (d,1H,J = 8.8 Hz),3.39 (s,3H),3.27 (t,1H,J = 6.4 Hz),2.65 (s,1H),2.56 (t,2H,J = 6.8 Hz),2.32-2.20 (m,2H),1.92-1.78 (m,2H),1.77-1.61 (m,2H),1.58-1.48 (m,1H),1.05 (d,3H,J = 7.2 Hz),0.89 (s,9H),0.83 (d,3H,J = 6.8 Hz),0.07 (s,6H); 13C NMR (100 MHz,CDCl3): δ 191.6,133.9,120.8,94.0,90.4,84.4,80.8,74.7,60.9,60.5,55.9,48.5,39.8,35.4,31.3,29.2,26.0,23.6,18.4,16.5,11.4,-5.2; HRMS (ES) m/z calcd. for C25H44O5SiNa (M+Na)+ 475.2856,found 475.2863.
Preparation of (8): To a solution of 7 (529.5 mg,1.17 mmol) in DCM (20 mL) was added NaHCO3 (491.5 mg,5.85 mmol) and Dess- Martin periodinane (992.4 mg,2.34 mmol) at 0℃. The reaction mixture was stirred at r.t. for 45 min,then quenched with saturated aqueous Na2SO3 (20 mL) and saturated aqueous NaHCO3 (20 mL). The aqueous layer was extracted with DCM (2 × 50 mL). Then the combined organic layers were dried over MgSO4,filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (10: 1,PE/EtOAc) to provide 8 (451.5 mg,85%) as a pale yellow oil. [a]D 27 -78.9 (c 0.4,CHCl3); IR (thin film,cm-1): 3442,2932,2925,2211,1716,1661,1383,1256,1155,1107,1027,920,833,779; 1H NMR (400 MHz,CDCl3): δ 5.72 (ddd,1H,J = 17.2,10.2,7.0 Hz),5.44 (d,1H,J = 17.2 Hz),5.34 (d,1H,J = 10.2 Hz),4.71 (d,1H,J = 6.8 Hz),4.62 (d,1H,J = 6.0 Hz),4.61 (d,1H,J = 6.8 Hz),3.73 (t,2H,J = 6.8 Hz),3.37 (s,3H),3.20 (t,1H,J = 6.4 Hz),3.02 (dq,1H,J = 10.8,7.2 Hz),2.52 (t,2H,J = 6.8 Hz),2.40-2.21 (m,2H),1.90-1.80 (m,2H),1.79-1.70 (m,1H),1.53-1.39 (m,1H),1.08 (d,3H,J = 7.2 Hz),1.03 (d,3H,J = 7.2 Hz),0.88 (s,9H),0.06 (s,6H); 13C NMR (100 MHz,CDCl3): δ 211.4,191.4,132.6,120.6,94.5,91.0,84.4,81.3,60.9,60.0,56.0,45.5,44.4,40.0,31.0,28.1,25.9,23.5,18.4,17.1,16.5,-5.2; HRMS (ES) m/z calcd. for C25H42O5SiNa (M+Na)+ 473.2699,found 473.2705.
Preparation of (9): To a suspension of CuI (117.6 mg,0.61 mmol) in THF (35 mL) at -78℃ was added MeMgBr (3.0 mol/L in Et2O,0.8 mL,2.46 mmol). The solution was stirred for 0.5 h at this temperature. A solution of 8 (191.2 mg,0.42 mmol) in THF (5 mL) was added dropwise to this solution and the reaction mixture was stirred at -78℃ for 5 min. The reaction was quenched with MeOH (1.0 mL),then poured into saturated aqueous NH4Cl (50 mL) and the resulting mixture was stirred for 1.5 h at r.t. (open to air),the reaction mixture turned to blue,The aqueous layer was extracted with EtOAc (2 × 50 mL). Then the combined organic layers were dried over Na2SO4,filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (20:1,PE/EtOAc) to provide 9 (152.7 mg,78%) as a colorless oil. [a]D 28 -67.7 (c 0.1,CHCl3); IR (thin film,cm-1): 3442,2929,1618,1384,1256,1094,1029,831; 1H NMR (400 MHz,CDCl3): δ 5.66 (ddd,1H,J = 17.2,10.4,8.8 Hz),5.27 (d,1H,J = 10.4 Hz),5.20 (d,1H,J = 17.2 Hz),4.64 (d,1H,J = 6.8 Hz),4.55 (d,1H,J = 6.8 Hz),4.07 (d,1H,J = 8.8 Hz),3.84-3.76 (m,1H),3.73-3.65 (m,1H),3.38 (s,3H),2.75 (s,1H),2.66 (dd,1H,J = 10.8,6.8 Hz),2.50-2.38 (m,2H),2.38-2.30 (m,1H),2.21-2.13 (m,1H),2.12 (s,3H),1.92-1.83 (m,1H),1.69-1.65 (m,1H),1.56-1.46 (m,2H),1.45-1.37 (m,1H),0.99 (d,3H,J = 6.6 Hz),0.96 (d,3H,J = 7.1 Hz),0.87 (s,9H),0.04 (d,6H,J = 2.4 Hz); 13C NMR (100 MHz,CDCl3): δ 208.6,141.6,140.3,135.3,120.8,94.4,83.7,79.6,63.4,57.7,56.0,43.7,41.8,40.2,34.4,33.6,29.2,26.1,21.6,18.5,15.7,
12.8,-5.1,-5.2; HRMS (ES) m/z calcd. for C26H46OSiNa (M+Na)+ 489.3012,found 489.3021.
Preparation of (10): To a stirred solution of P(OEt)3 (1.3 mL,7.8 mmol) in THF (30 mL) at -78℃ was added KHMDS (1.0 mol/L in THF,3.9 mL,3.9 mmol) under oxygen atmosphere. The solution was stirred for 0.5 h at this temperature. A solution of 9 (179.9 mg,0.4 mmol) in THF (5 mL) was added dropwise to this solution and the reaction mixture was stirred at -78℃ for 5 min. The reaction was quenched with NaHCO3 (150 mL),the aqueous layer was extracted with EtOAc (3 × 50 mL). Then the combined organic layers were dried over Na2SO4,filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel to provide the product as a colorless oil.
To a stirred solution of the product in DCM (70 mL) at -20℃ was added BF3.Et2O (0.25 mL,2.0 mmol). The solution was stirred for 10 min at this temperature. The reaction was quenched with NaHCO3 (100 mL),the aqueous layer was extracted with EtOAc (2 × 70 mL). Then the combined organic layers were dried over Na2SO4,filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (6: 1,PE/EtOAc) to provide 10 (86.1 mg,86% over two steps) as a colorless oil. [a]D 30 73.4 (c 0.13,CHCl3); IR (thin film,cm-1): 3437,2962,2926,2873,1768,1664,1383,1150,1025,937,822; 1H NMR (400 MHz,CDCl3): δ 5.78 (ddd,1H,J = 16.8,10.0,8.0 Hz),5.47 (d,1H,J = 16.8 Hz),5.35 (d,1H,J = 10.0 Hz),5.28 (d,1H,J = 8.0 Hz),3.87 (ddd,1H,J = 15.6,8.0,4.0 Hz),3.71 (ddd,1H,J = 15.6,8.0,4.0 Hz),3.45 (s,1H),2.86 (t,1H,J = 5.2 Hz),2.33-2.23 (m,2H),2.23-2.15 (m,2H),2.13 (ddd,1H,J = 14.8,8.0,4.0 Hz),2.04 (ddd,1H,J = 14.8,8.0,4.0 Hz),1.92-1.90 (m,1H),1.77-1.68 (m,1H),1.50 (s,3H),1.44-1.38 (m,1H),1.17 (d,3H,J = 6.8 Hz),0.89 (d,3H,J = 7.6 Hz); 13C NMR (100 MHz,CDCl3): δ 197.6,164.1,137.9,135.0,120.7,90.7,86.1,85.6,59.8,55.2,47.8,40.7,32.4,32.1,29.3,25.6,19.4,18.0; HRMS (ES) m/z calcd. for C18H26O4Na (M+Na)+ 329.1729,found 329.1735.
Preparation of (2): A pressure tube equipped with a stir bar was charged with 10 (11.5 mg,0.037 mmol) and Hoveyda-Grubbs Catalyst 2nd Generation (4.7 mg,0.007 mmol). A balloon of argon was utilized to blanket the flask with inert gas. Under argon,the flask was cooled to -78℃ and the isobutylene gas was introduced until ~5 mL of liquid was condensed. Swiftly,the argon balloon was exchanged for the screw-cap and the reaction was warmed to room-temperature and then heated at 40℃ for 30 h. The vessel was recooled to -78℃ to remove the cap,then allowed to warm to room temperature. Once the isobutylene is evaporated,the residue was dissolved in CH2Cl2 (1 mL) and directly subjected to silica gel chromatography (3:1,PE/EtOAc) to provide 2 (5.1 mg,45%) as a colorless oil. [a]D 23 15.0 (c 0.1,MeOH); IR (thin film,cm-1): 3437,2926,1618,1383,1050,822; 1HNMR (400 MHz,acetone-d6): δ 5.62 (dd,1H,J = 9.6,1.2 Hz),5.13 (d,1H,J = 9.6 Hz),4.13 (s,1H),3.70-3.53 (m,2H),3.35-3.22 (m,1H),2.39-2.29 (m,1H),2.20-2.10 (m,4H),2.03-1.98 (m,2H),1.81 (d,3H,J = 1.2 Hz),1.77 (d,3H,J = 1.2 Hz),1.69-1.64 (m,1H),1.45-1.39 (m,1H),1.37 (s,3H),1.12 (d,3H,J = 7.2 Hz),0.93 (d,3H,J = 7.2 Hz); 13CNMR(100 MHz,acetone-d6): δ 197.7,165.7,139.3,138.4,123.4,89.0,86.0,80.6,59.4,56.1,47.8,42.4,33.0,32.3,29.6,26.6,26.0,19.2,18.3(2 C);HRMS(ES) m/z calcd. for C20H30O4Na (M+Na)+ 357.2042,found 357.2039.
Preparation of (3): To a solution of 2 (3.0 mg,0.009 mmol) in DCM (2 mL) was added NaHCO3 (7.6 mg,0.090 mmol) and Dess- Martin periodinane (19.0 mg,0.045 mmol) at 0℃. The reaction mixture was stirred at r.t. for 2 h,then quenched with saturated aqueous Na2SO3 (10 mL) and saturated aqueous NaHCO3 (10 mL). The aqueous layer was extracted with DCM (3 × 10 mL). Then the combined organic layers were dried over MgSO4,filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (5:1,PE/EtOAc) to provide 3 (3.0 mg,quant) as a colorless oil. [a]D 23 -23.0 (c 0.1,MeOH); IR (thin film,cm-1): 3443,2924,2854,1725,1664,1461,1380,1262,1078,1022,800; 1H NMR (400 MHz,acetone-d6): δ 9.73 (t,1H,J = 2.8 Hz),5.70 (dd,1H,J = 10.0,0.8 Hz),5.15 (dt,1H,J = 10.0,1.2 Hz),4.18 (s,1H),2.76 (dd,1H,J = 15.4,2.8 Hz),2.69 (dd,1H,J = 15.4,3.2 Hz),2.41-2.32 (m,1H),2.19-2.11 (m,4H),1.82 (d,3H,J = 1.2 Hz),1.77 (d,3H,J = 0.8 Hz),1.73-1.65 (m,1H),1.47 (s,3H),1.44-1.39 (m,1H),1.14 (d,3H,J = 6.8 Hz),0.87 (d,3H,J = 7.2 Hz); 13C NMR (125 MHz,acetone-d6): δ 201.7,197.7,166.8,139.5,137.9,122.9,86.7,86.1,81.2,56.2,52.9,47.7,33.2,32.3,30.3,27.0,25.9,19.1,18.3,18.2; HRMS (ES) m/z calcd. for C20H28O4Na (M+Na)+ 355.1885,found 355.1891.
3. Results and discussionWe paid our attention to total synthesis of norleucosceptroids F and G starting from compound 4 achieved by our group previously [4b]. The alkynyl lithium,which was genernated by mixing n-BuLi with compound 5,reacted with compound 4 to furnish a labile intermediate. The intermediate reacted with TEMPO/PhI(OAc)2 to furnish the aldehyde 6. Asymmetric allylation with (-)-B-methoxydiisopinocamphenylborane gave compound 7,without any diastereomers detectable. After oxidation of 7-8,the reaction with CuI/MeMgBr to give the desired compound 9. Subsequently,we installed the tertiary alcohol at C-11 position with proper stereochemistry after treatment of 9 with base in the presence of triethyl phosphite and oxygen [4f]. The resulting compound is so instable that it was directly transformed to compound 10 via oxa- Michael cyclization-dehydration-deprotection cascade with boron trifluoride.
To expediently access the required trisubstituted alkene fragment,cross metathesis (CM) was attempted instead of our established ozonolysis-Wittig sequence. Thus,we first use compound 11,easily synthesized by compound 9,as the model substrate to probe the reactivity of the CM reaction. Interestingly,applying commonly used 2-methyl-2-butene in cross metathesis of compound 11 failed to afford the desired compound 12 in significant yield [5]. Either compound 13 or compound 14 was generated at 40-60℃ or 140℃ respectively in the presence of Moor Ru-based catalysts. We ascribed this unexpected reactivity to the steric repulsion between 2-methyl-2-butene and the dihydrofuran in 11,and the possible chelation of allylic ethereal oxygen with metal catalyst in transition state of cross metathesis. The rationale of the formation of compounds 13 and 14 is illustrated in Scheme 2. At the relatively low temperature (40-60℃),the less hindered transition state led to compound 13; at 140℃,metal-catalyzed alkene transfer of compound 13 might take place firstly and the resultant terminal alkene would be subjected to the common cross metathesis with 2-methyl-2-butene to afford compound 14. Cross metathesis between compound 11 and 2- propene resulted in the formation of 12 with moderated yield.

|
Download:
|
| Scheme 2.The formation of compounds 13 and 14. | |
{kind=link}
Crossmetathesis between compound 10 and 2-propene resulted in the formation of norleucosceptroid F (2) in 45% yield due to the lability of 10 under the reaction condition. Final Dess-Martin oxidation delivered norleucosceptroid G (3) smoothly. The characteristic data of our synthetic sample fits well with those reported by Li’s group.
4. ConclusionIn general,we accomplished total synthesis of norleucosceptroids F and G on the basis of our first total synthesis of leucosceptroid B. The current synthesis exhibits our synthetic strategy can be a general one accessible to different members in leucosceptroid and norleucosceptroid family.
AcknowledgmentsWe appreciate the financial support from the National Natural Science Foundation of China (Nos. 21322205,21321061,J1103315) and the Ministry of Education of China (No. 20130181110022). We also thank the comprehensive training platform of the Specialized Laboratory in the College of Chemistry at Sichuan University for compound testing.
| [1] | (a) S.H. Luo, Q. Luo, X.M. Niu, et al., Glandular trichomes of Leucosceptrum canum harbor defensive sesterterpenoids, Angew. Chem. Int. Ed. 49 (2010) 4471-4475; (b) S.H. Luo, L.H. Weng, M.J. Xie, et al., Defensive sesterterpenoids with unusual antipodal cyclopentenones from the leaves of Leucosceptrum canum, Org. Lett. 13 (2011) 1864-1867; (c) S.H. Luo, J. Hua, C.H. Li, et al., New antifeedant C20 terpenoids from Leucosceptrum canum, Org. Lett. 14 (2012) 5768-5771; (d) S.H. Luo, J. Hua, X.M. Niu, et al., Defense sesterterpenoid lactones from Leucosceptrum canum, Phytochemistry 86 (2013) 29-35; (e) S.H. Luo, J. Hua, C.H. Li, et al., Unusual antifeedant spiro-sesterterpenoid from the flowers of Leucosceptrum canum, Tetrahedron Lett. 54 (2013) 235-237; (f) S.H. Luo, C.L. Hugelshofer, J. Hua, et al., Unraveling the metabolic pathway in Leucosceptrum canum by isolation of new defensive leucosceptroid degradation products and biomimetic model synthesis, Org. Lett. 16 (2014) 6416-6419. |
| [2] | C.H. Li, S.X. Jing, S.H. Luo, et al., Peltate glandular trichomes of Colquhounia coccinea var. mollis harbor a new class of defensive sesterterpenoids, Org. Lett. 15 (2013) 1694-1697. |
| [3] | G.Z. Yue, X. Huang, B. Liu, Progress in the total syntheses oftrans-hydrindanecontaining terpenoids, Chin. J. Org. Chem. 33 (2013) 1167. |
| [4] | (a) J. Xie, Y. Ma, D.A. Horne, Asymmetric synthesis of the core structure of leucosceptroids A-D, J. Org. Chem. 76 (2011) 6169-6176; (b) X. Huang, L.Q. Song, J. Xu, et al., Asymmetric total synthesis of leucosceptroid B, Angew. Chem. Int. Ed. 52 (2013) 952-955; (c) C.L. Hugelshofer, T. Magauer, A general entry to antifeedant sesterterpenoids: total synthesis of (+)-norleucosceptroid A, (-)-norleucosceptroid B, and (-)- leucosceptroid K, Angew. Chem. Int. Ed. 53 (2014) 11351-11355; (d) C.L. Hugelshofer, T. Magauer, Total synthesis of the leucosceptroid family of natural products, J. Am. Chem. Soc. 137 (2015) 3807-3810; (e) C.L. Hugelshofer, T. Magauer, Strategies for the synthesis of antifeedant leucosceptroid natural products, Synlett 26 (2015) 572-579; (f) S. Guo, J. Liu, D.W. Ma, Total synthesis of leucosceptroids A and B, Angew. Chem. Int. Ed. 54 (2015) 1298-1301. |
| [5] | A.K. Chatterjee, T.L. Choi, D.P. Sanders, et al., A general model for selectivity in olefin cross metathesis, J. Am. Chem. Soc. 125 (2003) 11360-11370. |