 2015, Vol.26
2015, Vol.26 



Alzheimer’s disease (AD),a neurodegenerative disorder,is the most common form of dementia and no disease modification therapies are currently available. It is affecting more and more people worldwide and the gradual loss of mental and physical functions of the patients poses great emotional and economic threat to the families and is a financial burden on the society for long-term care. This disease is characterized pathologically by the presence of intracellular neurofibrillary tangles (NFTs) and extracellular amyloid plaques. According to the amyloid cascade hypothesis,accumulation of amyloid peptides (βb) in the brain, which is the main component of senile plaques,results in neuronal toxicity and cell death [ 1 ]. βb is produced from β-amyloid precursor protein ( β-APP) by the sequential cleavage of the β- and γ-secretase [ 2 ]. β-Secretase (also known as BACE1,memapsin-2 or Asp-2) has been considered an attractive therapeutic target for the treatment of AD [ 3 ].
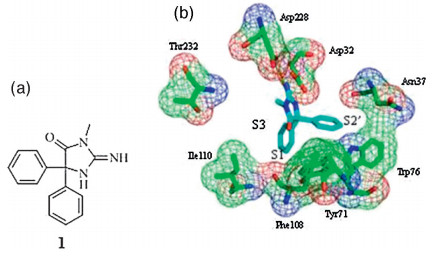
In this research we focused on a lead compound,2-imino-3- methyl-5,5-diphenylimidazolidin-4-one ( 1,Fig. 1). It is identified by several unique studies as a weak BACE1 inhibitor with an IC50 of 7.1 μmol/L [ 4, 5 ]. What makes this compound attractive is its ability to cross the blood-brain barrier (BBB),which precluded many excellent BACE1 inhibitors (like the HEA type,the first generation BACE1 inhibitor [ 6, 7 ] from further development. Based on this,a series of brain penetrable BACE1 inhibitors have been discovered [ 8 ],among which the most promising MK-8931 [ 9 ] has entered a phase 3 clinical trial.

|
Download:
|
| Fig. 1. Compound 1 (1a) and its interaction model with BACE1 active site (PDB:4DJU) (1b). | |
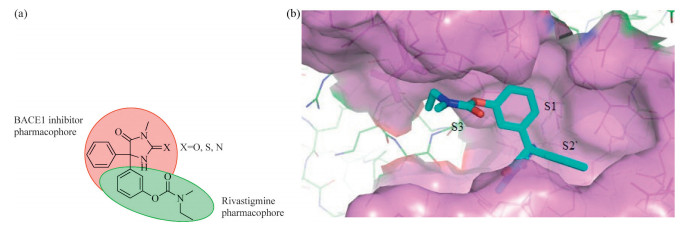
Taking compound 1 as a lead,we aimed at discovering new compounds with improved BACE1 inhibitory potency while maintaining its ability to cross the BBB. Analyzing the X-ray crystal structureofcompound 1 bindingtotheBACE1activesite(PDB:4DJU [ 10 ]) revealed that the H-bond interactions between the guanidine andAsp228and Asp32arecrucial foritsinhibitoryactivitywhile the two phenyl rings extended to the S1 and S2 0 pockets,respectively, leaving the S3 pocket unoccupied. Therefore,it might be possible to increase the inhibitory activity against BACE1 if a hydrophobic group is attached at the meta-position of one phenyl ring to extend to the S3 pocket. Based on this hypothesis,compounds described in Fig. 2a were designed. The phenolic ester group was introduced to bind totheS3pocket.Another reasonforthis designwasthat itisthe pharmacophore of a brain penetrable AChE inhibitor Rivastigmine [ 11, 12 ],which is now the main therapeutic drug used for AD. We hope that the integration of two brain penetrable pharmacophores would retain the brain accessibility while increasing the BACE1 inhibitory activity. We also want to replace the guanidine with urea or thiourea,as they are also capable of forming the essential H-bond interactions but the polarity of the molecule was greatly reduced. The reduction of the molecule polarity might help the brain penetration.One target compound (X = Natom)wasdockedinto the BACE1 active binding site to validate this design and the docking results confirmed our hypothesis above (Fig. 2b).

|
Download:
|
| Fig. 2. Designed compounds (2a, type I) and docking result of compound 11 with BACE1 active binding site (2b). | |
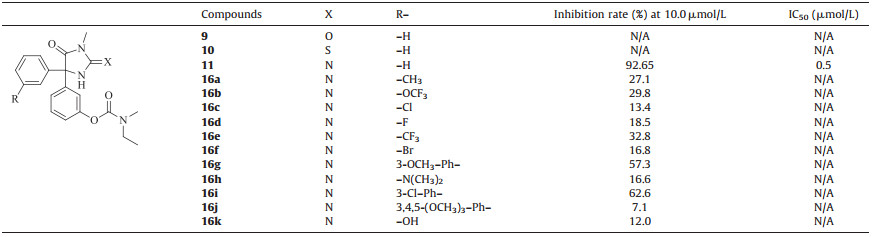
The synthesis of designed compounds 9,10 and 11 were shown in Scheme 1. 3-Hydroxybenzoic acid was used as the starting material. The phenol group was protected by reacting with CH3I in the presence of K2CO3 to give 3. Intermediate 4 was obtained by the hydrolysis of 3 with 4 mol/L NaOH (100% yield). The 3- methoxybenzoyl chloride prepared from 4 by reacting with SOCl2 under reflux underwent an AlCl3 catalyzed Friedel-Crafts reaction with benzene to afford 5 in a total yield of 77%. Deprotection of the methyl group using AcOH/HBr under reflux gave 6 (yield 100%). The phenol group was then transformed to a phenol ester group by reacting with ethyl(methyl)carbamic chloride to give 7 in a yield of 70%. A mixture of 7,NaCN and (NH4)2CO3 in ethanol/H2O was heated to reflux to give 8 (yield 54%). Target compound 9 was obtained by methylation of 8 with CH3I,acetone and K2CO3 in a yield of 48%. Lawesson’s reagent was used to transform the C55O group of9 to C=S to afford 10 (yield 63%). The C=S group of 10 was finally converted to C55N bond with tert-butyl hydroperoxide and ammonium hydroxide to give 11 in a yield of 21%. The synthesis of designed compounds 16a-16k were shown in Scheme 2. 3-Iodophenol was converted to 12 by reacting with ethynyltrimethylsilane under the catalysis of CuI/PdCl2(PPh3)2/ TEA (yield 100%). CuI/PdCl2(PPh3)2/TBAF catalyzed cross coupling of12 with 1-bromo-3-fluorobenzene (take R = -F as an example) at 80 ℃ resulted in 13 in a yield of 100%. 14 was obtained by the oxidation of the ethynyl group with KMnO4 in the presence of MgSO4 and Na2CO3 (yield 65%). Cyclization and rearrangement happened between 14 and 1-methylguanidine hydrochloride at 90 ℃ giving 15 in a yield of 58%. 15 reacted with ethyl (methyl) carbamic chloride to give the final product 16 (yield 24%). The structure of the new compounds was characterized by 1H NMR and MS.
|
|
Download:
|
| Scheme 1. Synthetic route of the designed compounds (type I).Reagents and conditions:(a) K2CO3,CH3I,r.t.;(b) NaOH,r.t.;(c) i.SOCl2,reflux ii.AlCl3,benzene,r.t.;(d) AcOH, 48% HBr,reflux;(e) ethyl (methyl) carbamic chloride,acetone,K2CO3,r.t.;(f) NaCN,(NH4)2CO3,120℃;(g) acetone,K2CO3,CH3I,r.t.;(h) Lawesson's reagent,reflux;(i)t-BuOOH, NH3·H2O,r.t. | |

|
Download:
|
| Scheme 2. Synthetic route of designed compounds (type II).Reagents and conditions:(a) PdCl2(PPh3)2,TEA,CuI,r.t.;(b) TBAF,PdCl2(PPh3)2,80℃;(c) KMnO4,MgSO4,Na2CO3, r.t.;(d)1-methylguanidine hydrochloride,Na2CO3,EtOH/H2O,90℃;(e) DIPEA,THF,reflux. | |
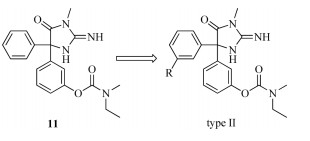
BACE1 inhibitor research has long been stagnating for not being able to cross the BBB until the discovery of compound 1. As an analogue of phenytion,it is brain penetrable and showed weak BACE1 inhibitory activity. This brings great hope to the BACE1 inhibitor research. In this report,14 analogues of compound 1 were designed and successfully synthesized. Compounds 9,10,11 were tested for their BACE1 inhibitory activity using the time-resolved fluorescence (TRF) method (Table 1). It was found that at a concentration of 10 μmol/L,9 and 10 exhibited almost no inhibitory activity against BACE1 while compound 11 showed an IC50 of 0.5 μmol/L,14-fold more potent than the lead compound 1,which is in accordance with our expectations. As the replacement of guanidine with urea and thiourea leads to a large bioactivity loss,it is suggested that the guanidine was essential for the BACE1 inhibitory activity. Unable to change the phenol ester group or the guanidine,we tried to introduce a group on the other phenyl ring to design another kind of compounds (type II, compounds 16a-16k) and investigate whether this modification further improves the potency (Fig. 3). Different type (electronwithdrawing or electron-donating) and size of groups were introduced. We hope that this modification could help enhancing the hydrophobic interactions between the small molecule and the protein active binding site thus improves the enzymatic potency.

|
Download:
|
| Fig. 3. Designed compounds (type II). | |
Compounds 16a-16k were also tested for their BACE1 inhibitory activity using the time-resolved fluorescence (TRF) method (Table 1). The results showed that inhibitory activity of these compounds (type II) against BACE1 was lower than that of compound 11. It suggests that when a hydrophobic group was attached to one phenyl of the lead compound 1 the other phenyl could not tolerate any substitutions. This is maybe due to the conformational changes induced by the meta- substitutions,making it difficult for the molecule to reach to the BACE1 binding site to form the interactions essential for its enzymatic activity. Efforts to optimize the structure of11 to further improve potency are ongoing.
| Table 1 Inhibitory activity of target compounds against BACE1. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In conclusion,based on the lead compound 1,a series of cyclic acylguanidine BACE1 inhibitors were designed and synthesized. Enzymatic inhibition tests showed that compound 11 exhibited a 14-fold improvement in potency,which implies that the designing strategy of extending the hydrophobic phenyl ester group from the S1 pocket to occupy the S3 pocket is of great value. This compound represents a good lead for the discovery of new BACE1 inhibitors.
AcknowledgmentsThis work was supported by grants from The National Natural Science Foundation of China (No. 81172924) and Beijing Municipal Natural Science Foundation (No. 7112106).
| [1] | J.A.Hardy,G.A.Higgins,Alzheimer's disease:the amyloid cascade hypothesis,Science 256(1992)184-185. |
| [2] | D.J.Selkoe,Translating cell biology into therapeutic advances in Alzheimer's disease,Nature 399(1999) A23-A31. |
| [3] | Y.Hamada,Y.Kiso,Recent progress in the drug discovery of non-peptidic BACE1 inhibitors,Expert Opin.Drug Discov.4(2009)391-416. |
| [4] | Z.N.Zhu,Z.Y.Sun,Y.Z.Ye,et al.,Discovery of cyclic acylguanidines as highly potent and selective β-site amyloid cleaving enzyme (BACE) inhibitors:Part I-inhibitor design and validation,J.Med.Chem.53(2010)951-965. |
| [5] | Z.N.Zhu,Iminoheterocycle as a druggable motif:BACE1 inhibitors and beyond,Trends Pharmacol.Sci.33(2012)233-240. |
| [6] | M.S.Wolfe,Secretase targets for Alzheimer's disease:identification and therapeutic potential,J.Med.Chem.44(2001)2039-2060. |
| [7] | R.Silvestri,Boom in the development of non-peptidic β-Secretase (BACE1) inhibitors for the treatment of Alzheimer's disease,Med.Res.Rev.29(2009)295-338. |
| [8] | D.Oehlrich,H.Prokopcova,H.J.M.Gijsen,The evolution of amidine-based brain penetrant BACE1 inhibitors,Bioorg.Med.Chem.Lett.24(2014)2033-2045. |
| [9] | J.D.Scott,A.W.Stamford,E.J.Gilbert,J.N.Cumming,Iminothiadiazine dioxide compounds as BACE inhibitors,compositions and their use,WO2011044181A1. |
| [10] | J.N.Cumming,E.M.Smith,L.Y.Wang,et al.,Structure based design of iminohydantoin BACE1 inhibitors:identification of an orally available,centrally active BACE1 inhibitor,Bioorg.Med.Chem.Lett.22(2012)2444-2449. |
| [11] | J.Corey-Bloom,R.Anand,J.Veach,A randomized trial evaluating the efficacy and safety of ENA 713(rivastigmine tartrate),a new acetylcholinesterase inhibitor,in patients with mild to moderately severe Alzheimer's disease,Int.J.Geriatr.Psychopharmacol.1(1998)55-65. |
| [12] | S.I.Finkel,Effects of rivastigmine on behavioral and psychological symptoms of dementia in Alzheimer's disease,Clin.Therap.26(2004)980-990. |