 2015, Vol.26
2015, Vol.26 


 , Qing-Qing Yina, Li-Juan Zhanga, Hua-Chuan Zhengb, Hong-Rui Songa
, Qing-Qing Yina, Li-Juan Zhanga, Hua-Chuan Zhengb, Hong-Rui Songa
b The First Affiliated Hospital of Liaoning Medical University, Jinzhou 121001, China
The Hedgehog (Hh) protein family,which includes Sonic (Shh),Indian (Ihh),and Desert (Dhh) hedgehogs,regulated cell growth and migration during embryonic development [1, 2, 3]. Activation of the Hh signaling pathway is initiated by Shh ligands bound to its receptor Patched (Ptch),which relieves its inhibition of Smoothen (Smo) receptor. Smo activation triggers a series of intracellular events ultimately leads to specific gene expression mediated by the Gli family transcription factors [4, 5]. Hh signaling pathway was normally silent in adult tissues,nevertheless aberrant activation of the Hh pathway was associated with certain cancers. Thus,the blockade of Hh pathway had been investigated as a novel strategy in cancer chemotherapy [6, 7].
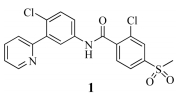
Inhibition of Smo activity has shown some promise in the treatment of cancers driven by activating mutations of the Hh pathway [8, 9, 10]. Furthermore,several Hh pathway antagonists have proceeded to clinical development,among which vismodegib (1,Fig. 1) has obtained marketing authorization in the United States in 2012 [11, 12]. Sonidegib (2,Fig. 2),a clinical stage Hh inhibitor developed by Novartis,is awaiting for the registration in the U.S. for the treatment of patients with advanced basal cell carcinoma. Sonidegib (2) bearing a morpholinopyridine unit suppressed the growth of Hh pathway-dependent tumors by selective inhibition of the positive regulator smoothened (Smo) [13, 14]. LEQ506 (3a),a second-generation Hh inhibitor,is currently being investigated in a Phase I clinical trial for patients with solid tumors. SAR studies had demonstrated the replacement of the benzylic methylene linker with an oxygen atom (3b) was well tolerated,whereas replacement with an NH group (3c) resulted in a 10-fold decrease in inhibition of the Hh pathway [15].

|
Download:
|
| Fig. 1.The structure of vismodegib (1). | |
Inspired by the structural characteristics of Sonidegib (2) and LEQ506 (3a),we envisioned that the merging of these two bioactive components would afford a hybrid structure with the potential for antiproliferative activity. We therefore adopted the biaryl ether (Part A) active fragment from LEQ506 analog (3b) and morpholino pyridine (Part B) unit from Sonidegib (2) in target compounds,and a carbonyl group or an amide group was selected as the linker between the two parts. Additionally,the order of heteroaryl or aryl cycloaliphatic amine units in the target compounds attaching to the linker was reversed based on the functional group reversion principle. Eventually,a series of 4- substituted-phenoxy-benzamide derivatives (Ⅰ,Ⅱ Fig. 2) were obtained. In this paper,the synthesis of these benzamide derivatives was reported,and their biological activities were evaluated.

|
Download:
|
| Fig. 2. Sonidegib (2), LEQ506 analogs (3a–c) and general structure of the target compounds (I and II). | |
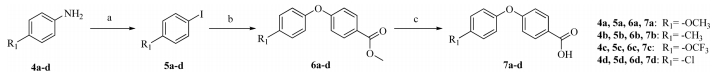
The key intermediates 7a-d were synthesized according to the routes outlined in Scheme 1. Aryl iodides were synthesized from aryl amines by a diazotization reaction. The generated diazonium salts were iodinated with KI to give compounds 5a-d [16]. The substituted iodobenzenes 5a-d were coupled to methylparaben to afford compounds 6a-d,via an N,N-dimethyl glycine-promoted Ullmann coupling. Compounds 6a-d were further hydrolyzed to compounds 7a-d in excellent yields under reflux [17].

|
Download:
|
| Scheme. 1. Reagents and conditions: (a) (1) NaNO2, H2SO4, 0 ℃, 2 h; (2) KI, dichloromethane, 0 ℃, 6 h; (b) 4-methyl-1-iodobenzene, CuI, N,N-dimethylglycine, Cs2CO3, 1,4-dioxane, reflux under nitrogen atmosphere, 24 h; (c) NaOH, ethanol solution, reflux, 3 h. | |
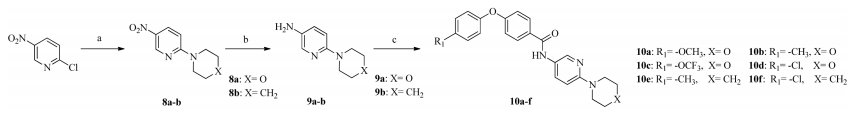
Compounds 10a-f were synthesized according to the routes outlined in Scheme 2. 2-Chloro-5-nitropyridine was converted to compounds 8a and b in the presence of morpholine or piperidine [18]. The resulting nitro compounds 8a and b were reduced to the amino compounds 9a and b using stannous chloride dihydrate and hydrochloric acid in aqueous ethanol,and then alkalized with sodium hydroxide solution [19]. 7a-d were treated with thionyl chloride to produce the corresponding acyl chloride. 9a and b reacted with acyl chloride to give the target compounds 10a-f [20].

|
Download:
|
| Scheme. 2.Reagents and conditions: (a) morpholine or piperidine, K2CO3, THF, reflux, 4 h; (b) SnCl2-2H2O, ethanol, HCl, reflux, 8 h; (c) acyl chloride of 7a–d, Et3N, dichloromethane, 0 ℃, 12 h. | |
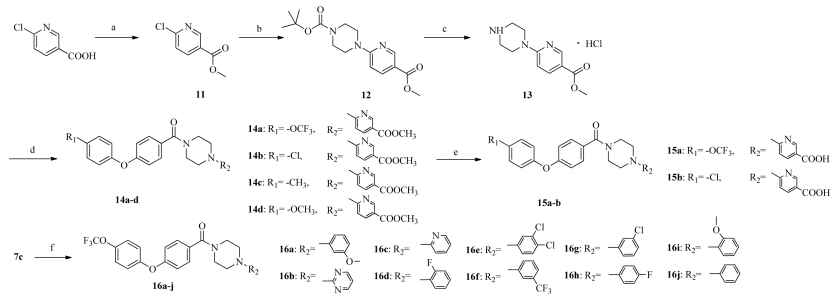
Compounds 14a-n,15a-b,16a-j were synthesized according to the routes outlined in Scheme 3. Compound 11 was synthesized from 6-chloronicotinic acid via an esterification reaction and then converted to compound 12 using a substitution reaction [21]. Compound 12 was deprotected to give compound 13 under acid conditions [22]. Intermediate 13 was reacted with the corresponding acyl chlorides 7a-d to give the target compounds 14a-d,which were further hydrolyzed to compounds 15a and b. Similarly,compounds 16a-j were obtained by the reaction of the acyl chloride 7c and various aryl piperazine [23].

|
Download:
|
| Scheme. 3.Reagentsand conditions:(a) Methanol,concentrated sulfuric acid,65 ℃; (b) 1-boc-piperazine,K2CO3, DMAP,DMF, 110 ℃,24 h; (c)concentrated hydrochloric acid in dioxane, dichloromethane 30 ℃, 2 h; (d) chloride of 7a–d, Et3N, dichloromethane, 0 ℃, 4 h; (e) NaOH, ethanol solution, reflux, 3 h; (f) chloride of 7a–d, aryl piperazidine or heteroaryl piperazidine, Et3N, dichloromethane, 0 ℃, 4 h. | |
General procedure for preparation of target compounds was given in the Supporting information.
3. Results and discussionAll the target compounds (10a-f,14a-d,15a-b and 16a-j) were evaluated for their antiproliferative activity against human colorectal cancer cell lines (SW620 and HT29),and human gastric cancer cell line (MGC803) using the MTT assay with vismodegib as a positive control. The results expressed as half maximal inhibitory concentration (IC50) values are summarized in Table 1.
| Table 1 In vitro cell growth inhibition by the target compounds and vismodegib (1). |
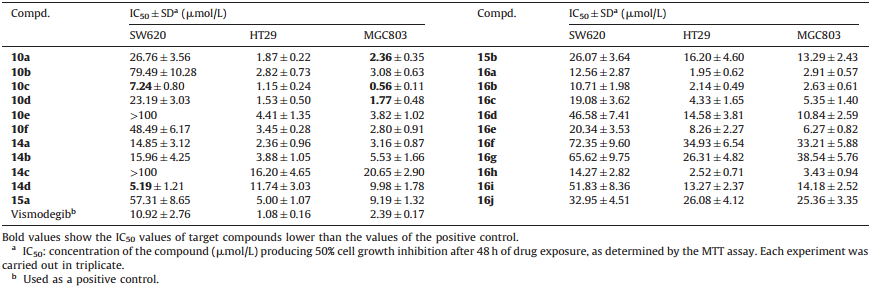
Initially,target compounds were divided into two regions: diaryl ether (Part A),aryl cycloaliphatic amine moiety (Part B). In general,most of them displayed high efficacy in HT29 and MGC803 cell lines. At the outset,our focus was on the modifications of Part A,including the substitutions on the para position of phenyl. The para-trifluoromethoxy substituent 10c (IC50 = 1.15 μmol/L [HT29],IC50 = 0.56 μmol/L [MGC803]) showed decent activity,however,para-methoxy and para-chlorine led to a significant loss of activity (10c vs 10a,10d or 14a vs 14b,14d) against MGC803 cells,confirming the beneficial impact of trifluoromethoxy in the R1 position.
Further investigations focusing on Part B on the antiproliferative activity were performed. On comparing 10d with 10f,it was found that morpholino pyridine surrogates were superior to piperidyl pyridine surrogates. Through functional group reversion,pyridyl piperazidine analog 16c (IC50 = 4.33 μmol/L [HT29],IC50 = 5.35 μmol/L [MGC803]) was obtained and exhibited moderate potencies. Addition of a polar methoxycarbonyl (14a,IC50 = 2.36 μmol/L [HT29]) group to pyridine increased the activity while its hydrolysate 15a lost potency by nearly two-fold. When the pyridyl was changed to a pyrimidyl (16b,IC50 = 2.14 μmol/L [HT29]),a two-fold increase of activity was observed. However,the trend did not hold in other analogs,as unsubstituted phenyl piperazidine analog 16j was less potent. Attempts to increase the antiproliferative activity were made by adding various groups to the phenyl ring (Part B). It was worth noting that the para-fluorine (16h,IC50 = 2.52 μmol/L [HT29]) and meta-methoxy (16a,IC50 = 1.95 μmol/L [HT29]) analogs were much more active than other substituted phenyl piperazidine analogs. Overall,the structure-activity relationships (SAR) study revealed that the antiproliferative activity of 3 compounds (10a,10c,10d) was comparable to that of the positive control vismodegib (1) in HT29 and MGC803 cells.
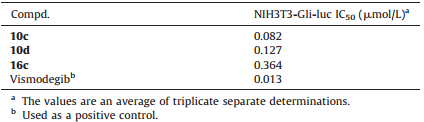
As shown in Table 2,the selected compound 10c,10d,16c were further tested for their Hh signaling inhibition using a luciferase reporter assay [24, 25] in NIH3T3 cells carrying a stably transfected Gli-reporter construct (NIH3T3-Gli-luc reporter cell line). This assay can effectively identify hedgehog signaling pathway inhibitors. It was suggested that compound 10c (IC50 = 0.082''' μmol/L) was less active as compared to the positive control vismodegib (IC50 = 0.013 μmol/L). In addition,10c and 10d (IC50 = 0.127 μmol/L) showed higher potency than 16c (IC50 = 0.364 μmol/L) in parallel to their antiproliferative activity. Although the hedgehog pathway inhibition of 10c was less potent than that of vismodegib,these results were encouraging and worthy of further investigation owing to the large structural changes involved.
| Table 2 The hedgehog pathway inhibition of compound 10c, 10d, 16c and vismodegib (1). |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
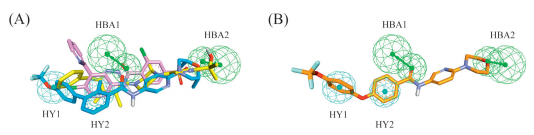
Then we examined if compound 10c fit the proposed pharmacophoric model for Hh antagonists. A conformational analysis was performed using the Common feature program in the Discovery studio 3.0 software. This analysis was conducted on the three compounds covering conformers with a range of 20 kcal/mol with respect to the global minimum that was used in building the best pharmacophoric model. According to this,the low energy conformers of vismodegib (1),sonidegib (2) and LEQ506 (3a) showed a very similar orientation and fulfilled all the pharmacophoric features of the model.
This model was built up by two hydrogen bond acceptor (HBA1- 2) groups and two hydrophobic (HY1-2) regions. Fitting of vismodegib (1),sonidegib (2) and LEQ506 (3a) to the pharmacophoric model was shown in Fig. 3A. Analysis of the superposition pattern of 10c showed a good fit between the molecule and the pharmacophoric model (Fig. 3B). For instance,the carbonyl oxygen in the amide moiety of compound 10c matched HBA1,and the oxygen atom in the morpholine moiety (Part B) corresponded to the HBA2 feature of the model. Moreover,the two phenyl rings in the diaryl ether moiety (Part A) were superimposed to the hydrophobic regions HY1 and HY2 perfectly.

|
Download:
|
| Fig. 3.(A) Pharmacophore models for vismodegib, sonidegib and LEQ506. Graphical representation of vismodegib (pink), sonidegib (blue), LEQ506 (yellow) fitted to the proposed pharmacophoric model for hedgehog antagonists. Pharmacophoric features are color coded: green for hydrogen bond acceptor groups (HBA1-2) and cyan for hydrophobic regions (HY1-2). HBA features are constituted by a smaller sphere accommodating the hydrogen bond acceptor group, by a directionality vector represented by an arrow, and by a larger sphere intended to allocate the hydrogen bond donor group of the target macromolecule. (B) Pharmacophore model for 10c. The atoms are color coded: Orange, carbon; white, hydrogen; red, oxygen; blue, nitrogen. | |
{kind=link}
In summary,a series of novel and distinctive 4-substitutedphenoxy-benzamide analogs were synthesized and characterized. Furthermore,three human cancer cell lines were used to evaluate their antiproliferative activity; the majority of analogs exhibited moderate activity and high selectivity in HT29 and MGC803 cell lines. In particular,the most promising compound 10c (Hh pathway inhibition IC50 = 0.082 μmol/L) displayed desirable antiproliferative activity with IC50 values of 1.15 μmol/L and 0.56 μmol/L against HT29 and MGC803 cell lines,respectively. The analysis of SAR indicated that compounds with a paratrifluoromethoxyl group on Part A were more potent than those with other substituents. In addition,the morpholino pyridine fragment in Part B exhibited higher efficacy as compared to the aryl piperazidine fragment. Further structural optimization studies are presently in progress and will be reported in due course.
AcknowledgmentsThe work was supported by Program for Innovative Research Team of the Ministry of Education of China and Program for Liaoning Innovative Research Team in University. Authors wish to thank to the Innovation and entrepreneurship training program for college students in Liaoning Province (No. 201410163009).
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.06.017.
| [1] | J.A. Williams, O.M. Guicherit, B.I. Zaharian, et al., Identification of a small molecule inhibitor of the hedgehog signaling pathway:effects on basal cell carcinoma-like lesions, Proc. Natl. Acad. Sci. U.S.A. 100(2003) 4616-4621. |
| [2] | N. Mahindroo, C. Punchihewa, N. Fujii, Hedgehog-gli signaling pathway inhibitors as anticancer agents, J. Med. Chem. 52(2009) 3829-3845. |
| [3] | H. Onishi, M. Katano, Hedgehog signaling pathway as a therapeutic target in various types of cancer, Cancer Sci. 102(2011) 1756-1760. |
| [4] | M.K. Hadden, Hedgehog pathway inhibitors:a patent review (2009-present), Expert Opin. Ther. Pat. 23(2013) 345-361. |
| [5] | A. Pimentel, M. Velez, L.J. Barahona, et al., New prospects for drug development:the hedgehog pathway revealed. Focus on hematologic malignancies, Future Oncol. 9(2013) 681-697. |
| [6] | J.M. Ruch, E.J. Kim, Hedgehog signaling pathway and cancer therapeutics:progress to date, Drugs 73(2013) 613-623. |
| [7] | C. Dockendorff, M.M. Nagiec, M. Weïwer, Macrocyclic hedgehog pathway inhibitors:optimization of cellular activity and mode of action studies, ACS Med. Chem. Lett. 3(2012) 808-813. |
| [8] | S.X. Atwood, M. Li, A. Lee, J.Y. Tang, A.E. Oro, GLI activation by atypical protein kinase C ı/λ regulates the growth of basal cell carcinomas, Nature 494(2013) 484-488. |
| [9] | Y. Wang, A.C. Arvanites, L. Davidow, et al., Selective identification of Hedgehog pathway antagonists by direct analysis of smoothened ciliary translocation, ACS Chem. Biol. 7(2012) 1040-1048. |
| [10] | F. Manetti, H. Faure, H. Roudaut, et al., Virtual screening-based discovery and mechanistic characterization of the acylthiourea MRT-10 family as smoothened antagonists, Mol. Pharmacol. 78(2010) 658-664. |
| [11] | S.E. Gould, J.A. Low, J.C. Marsters, et al., Discovery and preclinical development of vismodegib, Expert Opin. Drug Discov. 9(2014) 969-984. |
| [12] | A.E. Proctor, L.A. Thompson, C.L. O'Bryant, Vismodegib:an inhibitor of the hedgehog signaling pathway in the treatment of basal cell carcinoma, Ann. Pharmacother. 48(2014) 99-106. |
| [13] | S.F. Pan, X. Wu, J.Q. Jiang, et al., Discovery of NVP-LDE225, a potent and selective smoothened antagonist, ACS Med. Chem. Lett. 1(2010) 130-134. |
| [14] | M. Zollinger, F. Lozaćh, E. Hurh, et al., Absorption, distribution, metabolism, and excretion (ADME) of 14C-sonidegib (LDE225) in healthy volunteers, Cancer Chemother. Pharmacol. 74(2014) 63-75. |
| [15] | S. Peukert, F. He, M. Dai, et al., Discovery of NVP-LEQ506, a second-generation inhibitor of smoothened, Chem. Med. Chem. 8(2013) 1261-1265. |
| [16] | A.R. Hajipour, M. Seddighi, Application of[Hcpy] HSO4 brönsted acidic ionic liquid for the synthesis of aryl iodides from aromatic amines, Org. Prep. Procd. Inter. 43(2011) 292-296. |
| [17] | Y.H. Yang, Z.L. Wang, J.Z. Yang, et al., Design, synthesis and evaluation of novel molecules with a diphenyl ether nucleus as potential antitubercular agents, Bioorg. Med. Chem. Lett. 22(2012) 954-957. |
| [18] | H.W. Ding, Z. Chen, C.L. Zhang, et al., Synthesis and cytotoxic activity of some novel N-pyridinyl-2-(6-phenylimidazo[2,1-b]thiazol-3-yl) acetamide derivatives, Molecules 17(2012) 4703-4716. |
| [19] | B. Yang, A.W. Hird, D.J. Russell, et al., Discovery of novel hedgehog antagonists from cell-based screening:isosteric modification of p38 bisamides as potent inhibitors of SMO, Bioorg. Med. Chem. Lett. 22(2012) 4907-4911. |
| [20] | Y.B. Chen, J.L. Li, X.S. Shao, et al., Design, synthesis and insecticidal activity of novel anthranilic diamides with benzyl sulfide scaffold, Chin. Chem. Lett. 24(2013) 673-676. |
| [21] | K.M. Jung, K.H. Kim, J.L. Jin, M.J. Cho, D.H. Choi, Deep-red light-emitting phosphorescent dendrimer encapsulated tris-[2-benzo[b]thiophen-2-yl-pyridyl] iridium(III) core for light-emitting device applications, J. Polym. Sci., A:Polym. Chem. 46(2008) 7517-7533. |
| [22] | J. Wehbe, V. Rolland, A. Fruchier, M.L. Roumestant, J. Martinez, Enantioselective synthesis of new 4-substituted glutamic acid derivatives, Tetrahedron:Asym. 15(2004) 851-858. |
| [23] | T. You, K. Chen, F.H. Wang, et al., Design, synthesis, and biological evaluation of Nhydroxycinnamamide/salicylic acid hybrids as histone deacetylase inhibitors, Chin. Chem. Lett. 25(2014) 474-478. |
| [24] | J.K. Chen, J. Taipale, K.E. Young, T. Maiti, P.A. Beachy, Small molecule modulation of Smoothened activity, Proc. Natl. Acad. Sci. U.S.A. 99(2002) 14071-14076. |
| [25] | M.H. Xin, L.D. Zhang, F. Tang, et al., Design, synthesis, and evaluation of pyrrolo[2,1-f] [1,2,4] triazine derivatives as novel hedgehog signaling pathway inhibitors, Bioorg. Med. Chem. 22(2014) 1429-1440. |