 2015, Vol.26
2015, Vol.26 



b Nanjing Gator MediTech Company, Ltd., Nanjing 211300, China
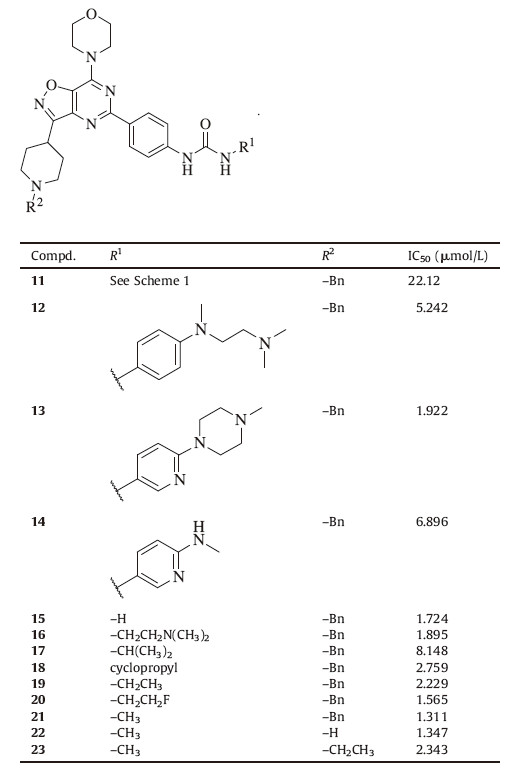
Phosphoinositide-3-kinases (PI3Ks) are members of lipid kinase family that phosphorylate inositol lipids at the 3-hydroxy group [1]. They play key regulatory roles in many cellular processes, including cell growth,survival,proliferation,motility,metabolism, and differentiation [2, 3, 4]. They are the central components of the PI3K/Akt/mTOR signaling pathway and activated by receptor tyrosine kinases (RTKs) and G-protein coupled receptors (GPCRs). The activated PI3Ks transduce signals from various growth factors and cytokines into intracellular messages by generating phospholipids. As the key second messengers,these phospholipids activate the serine/threonine protein kinase Akt and other downstream signaling pathways,promoting cell growth and proliferation [2]. PI3Ks can be divided into three classes according to their structures,substrates and functions,including class I (IA and IB),II and III. The class I are heterodimers composed of a regulatory subunit and a catalytic subunit. The class IA consists of p85 and p110 (α,β and δ),whereas the class IB is p101 and p110γ. The class II consists of a monomeric catalytic subunit,including three isoforms (PI3KC2α,PI3KC2β and PI3KC2γ). The class III consists of a single heterodimer of a catalytic (Vps34) and a regulatory (Vsp15) subunit [5, 6]. Aberrations of PI3K signaling are found in various tumors,including breast,prostate,colon,liver,and pancreatic cancers. The most frequently observed abnormalities are the loss or attenuation of PTEN function and mutations of the gene that encodes p110a [7]. The overexpression of p110β,δ,and γ has also been implicated in various forms of human tumors [4]. In addition,PI3K pathway activations also lead to resistance to current cytotoxic agents as well as targeted anticancer therapies [8]. Idelalisib (CAL-101 or GS-1101),a PI3Kδ inhibitor,is the first in its class that has gained the approvals of the FDA and EMA for the treatment of relapsed chronic lymphocytic leukemia (CLL) in July 2014. A number of other PI3K inhibitors are currently being studied in clinics,including GDC-0941,XL-147,BKM-120 and BEZ235 (Fig. 1) (http://ClinicalTrials.gov,[4]). These data indicate that regulating the activities of PI3Ks is an effective strategy for anticancer therapy.

|
Download:
|
| Fig. 1.Reported PI3K inhibitors. | |
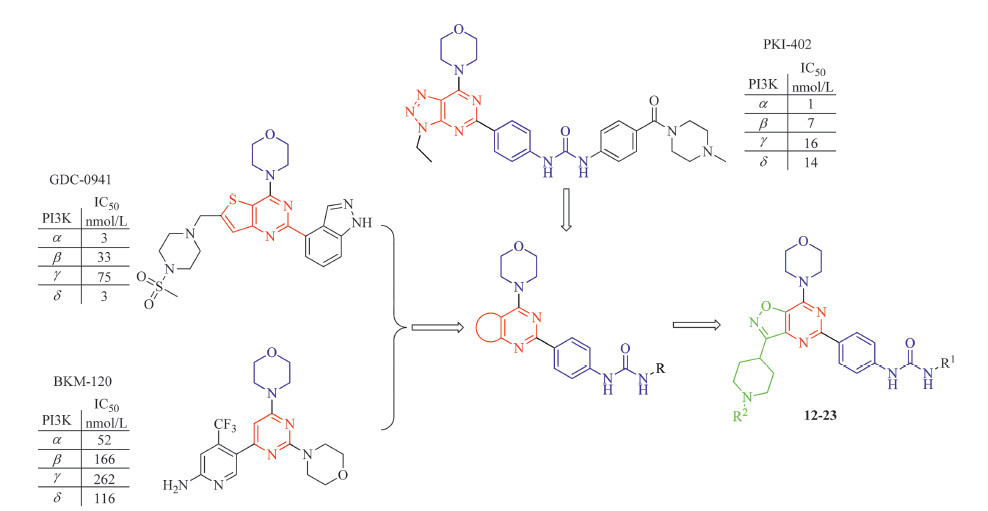
PI3K inhibitors from several structural classes,such as GDC- 0941,BKM-120,and PKI-402 [9] contain a common 2,4-disubstituted pyrimidine fragment with 4-morpholino group at the 4-position. Their binding data indicated that 2- and 4-substituents could pick up key hydrogen bond interactions with PI3Ks,whereas the pyrimidine ring mainly serve as a scaffold [9, 10, 11]. Based on this hypothesis,we designed the novel 3-(piperidin-4-yl)isoxazolo[ 4,5-d]pyrimidines (Fig. 2). This bicyclic template allows us to maintain the 2,4-disubstituented pyrimidine structure while investigating the effect of the fused 3-(piperidin-4-yl)isoxazole. This report describes the syntheses of a series of novel 3- (piperidin-4-yl)isoxazolo[4, 5, d]pyrimidine derivatives. Their anti- proliferative activities were evaluated in a cellular CCK-8 assay against breast cancer cell line BT-474. The lead compounds,20 and 21,were found to be potent inhibitors of PI3Kδ.

|
Download:
|
| Fig. 2.Design considerations. | |
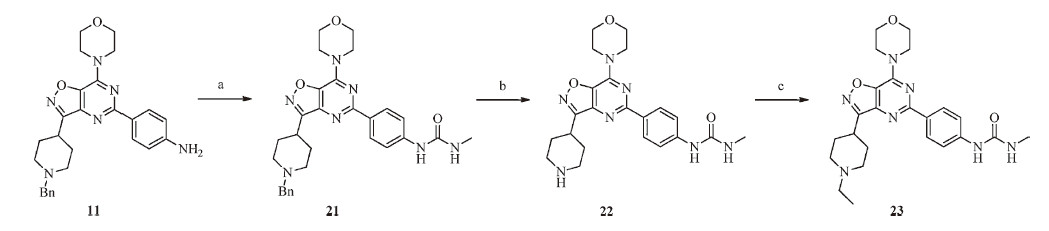
The syntheses of compounds 2-21 are outlined in Scheme 1. Details of reaction conditions and characterization data related to compounds 2-11 are provided in the Supplementary data [12, 13, 14, 15, 16, 17, 18, 19].
To a solution of N1-(2-(dimethylamino)ethyl)-N1-methylbenzene- 1,4-diamine (116 mg,0.6 mmol) and TEA (182 mg, 1.8 mmol) in anhydrous DCM (4 mL) at -18 ℃ was added dropwise a solution of triphosgene (89 mg,0.3 mmol) in anhydrous DCM (2 mL). After 15 min,a solution of compound 11 (94 mg,0.2 mmol) in anhydrous DCM (2 mL) was added. The mixture was stirred overnight and allowed to warm to room temperature. The reactionmixture was dilutedwith water (5 mL) and extracted with DCM(5 mL × 3). The combined organic layers were subsequently washed with H2O (8mL× 3) and brine (8 mL × 3),dried over anhydrous Na2SO4 and concentrated under vacuum. The crude product was purified on a silica gel column (DCM/MeOH/TEA = 60/1/1,v/v/v) to give compound 12 (50 mg, 36% yield) as a yellow solid.
General procedure for compounds 13-21: To a solution of compound 11 (94 mg,0.2 mmol) and TEA (61 mg,0.6 mmol) in anhydrous DCM (2 mL) at -18 ℃ was added dropwise a solution of triphosgene (30 mg,0.1 mmol) in anhydrous DCM (2 mL). After 15 min,a solution of amine (5 eq.) in anhydrous DCM (2 mL) or amine hydrochloride (5 eq.) and TEA (6 eq.) was added. The mixture was stirred overnight and allowed to warm to room temperature. The reaction mixture was diluted with water (5 mL) and extracted with DCM (5 mL × 3). The combined organic layers were subsequently washed with H2O (6mL× 3) and brine (6 mL × 3),dried over anhydrous Na2SO4 and concentrated under vacuum. The crude product was purified on a silica gel column or preparative thin-layer chromatography to give compounds 13-21 [20]. All data of compounds 12-21 are summarized in the Supplementary data.
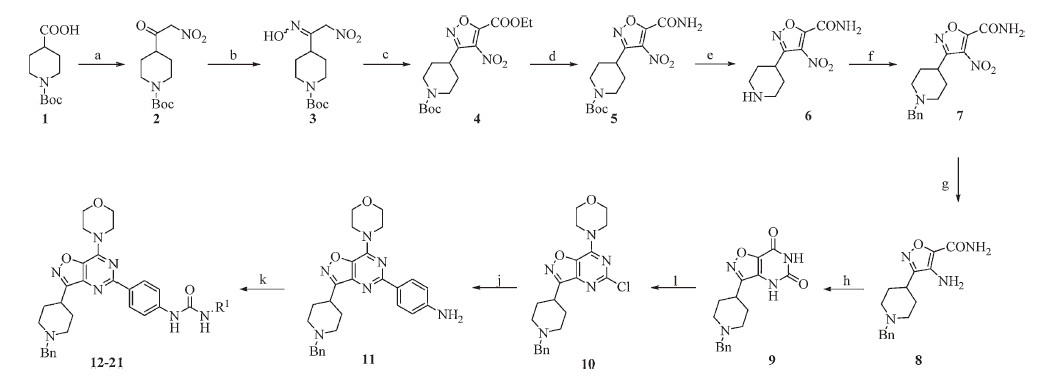
The syntheses of compounds 22 and 23 are outlined in Scheme 2. To a solution of compound 11 (471 mg,1 mmol) and TEA (126mg, 1.25mmol) in anhydrous DCM (4mL) was slowly added a solution of methylcarbamic chloride (103 mg,1.1mmol) in anhydrous DCM (3 mL). The mixture was then refluxed for 3 days. The reaction mixture was diluted with water (5mL) and extracted with DCM (8 mL × 3). The combined organic layers were subsequently washed with H2O (10mL× 3) and brine (10 mL × 3),dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified on a silica gel column (DCM/MeOH = 20/1,v/v) to give compound 21 (447 mg,71% yield) [21].

|
Download:
|
| Scheme. 2.Syntheses of compounds 22 and 23. Reagents and conditions: (a) Methylcarbamic chloride, TEA, anhydrous DCM, reflux, 3 d, 71%. (b) (1) ACE-Cl, DCE, r.t., overnight; (2) MeOH, reflux, 2 h; (3) (Boc)2O, TEA, DCM, 0 ℃, 3 h, 76%; (4) TFA, DCM, r.t., 2 h, 99% (di-TFA salt). (c) iodoethane, DIPEA, MeCN, -18 ℃-reflux, 69%. | |
To a solution of compound 21 (1.342 g,2.54 mmol) in DCE (40 mL) was added ACE-Cl (2.910 g,20.35 mmol) and stirred overnight at room temperature. The mixture was concentrated to dryness. The residue was refluxed in MeOH (20 mL) for 2 h and concentrated under vacuum to give the crude 1-methyl-3-(4-(7- morpholino-3-(piperidin-4-yl)isoxazolo[4, 5, d]pyrimidin-5-yl)phenyl) urea hydrochloride (1.218 g) [22]. This crude intermediate was suspended in DCM (50 mL) at 0 ℃. TEA (936 mg,9.25 mmol) and a solution of (Boc)2O (618 mg,2.83 mmol) in anhydrous DCM (10 mL) was then added. The mixture was stirred for 3 h,diluted with water (20 mL) and extracted with DCM (20 mL × 3). The combined organic layers were washed with H2O (25 mL × 3) and brine (25 mL × 3),dried over anhydrous Na2SO4,and concentrated in vacuo. The crude product was purified on a silica gel column (DCM/MeOH = 50/1,v/v) to give tert-butyl-4-(5-(4-(3-methylureido) phenyl)-7-morpholinoisoxazolo[4, 5, d]pyrimidin-3-yl)piperidine- 1-carboxylate (1.037 g,76% yield) as a white solid. The solution of the above intermediate (330 mg,0.61 mmol) in DCM (1 mL) and TFA (3 mL) was stirred for 2 h at room temperature. The mixture was concentrated to dryness in vacuo to give compound 22 as its di-TFA salt (405 mg,99% yield) as a yellow solid,which was used in the next step without further purification. Note: compound 22 was very difficult to purify ‘‘as is’’. The crystallization of its hydrochloride salt did not furnish a product with satisfactory yield and purity. The t-Boc-protection/deprotection steps were for the ease of purification only.
To a suspension of compound 22 di-TFA salt (100 mg, 0.15 mmol) and DIPEA (71 mg,0.55 mmol) in MeCN (3 mL) at -18 ℃ was added iodoethane (29 mg,0.18 mmol). The mixture was stirred for 1 h at the same temperature followed by stirring at room temperature for 5 h. TLC showed that reaction was not complete. The mixture was stirred at 65 ℃ for 11 h and followed by refluxing for 8 h. The reaction mixture was concentrated in vacuo and diluted with water (3 mL). The aqueous layer was adjusted to pH 14 using 6 mol/L NaOH (aq.) and extracted with DCM/ MeOH = 20/1 (5 mL × 3). The combined organic layers were dried over anhydrous Na2SO4. The crude product was purified on a silica gel column (DCM/MeOH/TEA = 55/1/1,v/v/v) to give compound 23 (48 mg,69% yield) as a yellow solid [16, 23]. All analytical data of compounds 22 and 23 were summarized in the Supplementary data. A small amount of quaternary salt was also observed during the reaction by LC-MS. It was easily removed during the work-up and purification.
2.2. Biological evaluationThe anti-proliferative activities of compounds 11-23 were assessed against BT-474 cells (human breast ductal carcinoma) by CCK-8 assay. BT-474 cells were plated in 96-well flat-bottomed microtiter plates (cells suspended in 100 μL culture medium per well) and incubated at 37 ℃ for 24 h under a 5% CO2 and 100% relative humidity atmosphere. A 25 μL aliquot of culture medium containing the tested compound was added to the wells. The plates were incubated for an additional 72 h. After removal of culture medium,fresh culture medium containing 10% CCK-8 was added and incubated at 37 ℃ for 2-4 h. The absorbance was measured on a SpectraMax M5 Microplate Reader at 450 nm. The percentage of inhibition at each compound concentration was calculated according to formula: The percentage of inhibition = (Asample - Anegative)/(Ablank - Anegative) × 100%. Compounds were studied for dose-response relationship at 100 μmol/L,25 μmol/L,6.25 μmol/ L,1.563 μmol/L,0.391 μmol/L,0.098 μmol/L,0.024 μmol/L, 0.006 μmol/L,0.0015 μmol/L,0.0004 μmol/L. Their IC50s were calculated using GraphPad Prism 5.
PI3Kδ kinase activity was determined using a PI3-Kinase (human) HTRFTM Assay kit in 384 well opaque black plates. CAL- 101 was used as a positive control. Typically,5 μL of ATP solution (40 μmol/L) was added into a mixture of 10 μL of PIP2 solution (20 μmol/L) containing PI3Kδ enzyme (80 ng) and 5 μL of test compound solution. The negative control and blank control were composed of the same mixed solutions except replacing test compound with DMSO. The blank control did not contain PI3Kδ enzyme. After a 30min incubation at room temperature,the reaction was stopped by the addition of 5 mL of stop buffer (stop A/ stop B = 3/1) followed by detection buffer (5 mL,DMC/DMA/ DMB = 18/1/1). After 1 h incubation at roomtemperature,emission signal was measured on EnVision® Multilabel Reader. Emission ratio (ER) of each well was calculated according to the formula: emission ratio (ER) = 665 nm emission signal/620 nm emission signal.The percentage of inhibitionat eachcompoundconcentration was calculated according to formula: The percentage of inhibition = (ERsample - ERnegative)/(ERblank - ERnegative) × 100%. Compounds were studied for dose-response relationship at 100 μmol/L, 25 μmol/L,6.25 μmol/L,1.563 μmol/L,0.391 μmol/L, 0.098 μmol/L,0.024 μmol/L,0.006 μmol/L,0.0015 μmol/L, 0.0004 μmol/L. Their IC50s were calculated using GraphPad Prism 5.
3. Results and discussion 3.1. ChemistryA facile synthesis of novel 3-(piperidin-4-yl)isoxazolo[4,5- d]pyrimidine scaffold was developed,starting from N-Boc-piperidine- 4-carboxylic acid (Scheme 1). Using this route,fifteen derivatives were synthesized for their anti-proliferative activity evaluation and initial structure activity relationship development.

|
Download:
|
| Scheme. 1.Syntheses of compounds 2–21. Reagents and conditions: (a) (1) CDI, anhydrous THF, N2, r.t., 1–2 h; (2) DBU, nitromethane, r.t., 36 h, 96%. (b) hydroxylamine hydrochloride, NaHCO3, EtOH, 50 ℃, overnight, 95%. (c) (1) ethyl 2-chloro-2-oxoacetate, anhydrous ether, r.t., 24 h; (2) TEA, 0 ℃-r.t., 60 h, 58%. (d) NH3/MeOH, r.t., 3 h, 96%. (e) TFA, DCM, r.t., 2 h, 100% (TFA salt). (f) BnBr, DIPEA, anhydrous MeCN, 0 ℃-r.t., 6 h, 81%. (g) Zn, NH4Cl, EtOH/water = 2/1, 0 ℃-r.t., 4 h, 78%. (h) triphosgene, anhydrous THF, reflux, 1 h, 90% (hydrochloride salt). (i) (1) POCl3, reflux, 24 h; (2) morpholine, TEA, DCM, -18 ℃, 10 min, 62%. (j) 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline, Pd(Pcy3)2Cl2, CsF, NMP/water = 9/1, Ar, 100 ℃, 48 h, 34%. (k) (1) TEA, triphosgene, anhydrous DCM, -18 ℃, 15 min; (2) amine or amine hydrochloride and TEA, -18 ℃-r.t., overnight, 30%–56%. | |
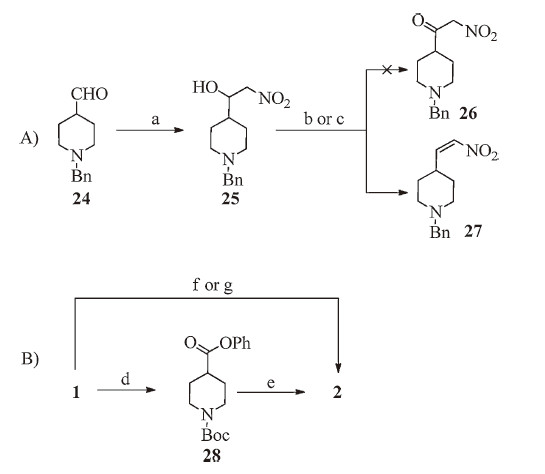
We at first attempted a two-step process for the synthesis of intermediate 26 as shown in Scheme 3A,which has the potential of avoiding the exchange of N-substitution on piperidine from Boc- to benzyl. Condensation of aldehyde 24 and nitromethane in the presence of KF worked well,giving the alcohol 25 in 96% yield. Oxidation of 25,under both Dess-Martin periodinane and Swern conditions,gave nitro olefin 27 in nearly quantitative yield (86% and 81%,respectively) instead of the α-nitroketone 26. These N-benzyl piperidine derivatives were also difficult to purify due to their good aqueous solubility. For the ease of intermediate isolation,we therefore decided to use the N-Boc-piperidines during the early part of the syntheses. Details of reaction conditions and characterization data related to compounds 24, 25 and 27 are summarized in the Supplementary data.

|
Download:
|
| Scheme. 3.Syntheses of a-nitroketone 2. Reagents and conditions: (a) Nitromethane, KF, 2-propanol, r.t., overnight, 96%. (b) Dess–Martin periodinane, anhydrous DCM, r.t., 35 min, 86%. (c) (1) oxalyl chloride, DCM, -78 ℃, DMSO, 15 min; (2) compound 25, DCM, -78 ℃, 1 h; (3) TEA, -78 ℃, 15 min, r.t., 1 h, 81%. (d) (1) (COCl)2, DMF, anhydrous DCM, 0 ℃, 2 h; (2) PhOH, TEA, anhydrous DCM, 0 ℃, 3 h, 43%. (e) Nitromethane, t-BuOK, DMSO, r.t., 8 h, 77%. (f) (1) CDI, anhydrous THF, N2, reflux, 1 h; (2) nitromethane, t-BuOK, reflux, 24 h, 75%. (g) CDI, anhydrous THF, N2, r.t., 1–2 h; (2) nitromethane, DBU, N2, r.t., 36 h, 96%. | |
As shown in Scheme 3B,key intermediate 2 can be obtained from N-Boc-piperidine-4-carboxylic acid (1) in a two-step process in moderate overall yield. The condensation of phenol ester 28 with nitromethane gave 2 in high yield (77%). However,the conversion of 1 to 28 in the presence of oxalyl chloride was only 43%. Alternatively,1 was first activated with CDI followed by addition of t-BuOK and nitromethane in anhydrous THF to give 2 in high overall yield (75%) in a one-pot procedure. Using DBU instead of t-BuOK as base,2 was obtained in excellent yield (96%). This optimized condition was used in the preparation of 3-(piperidin-4- yl)isoxazolo[4, 5, d]pyrimidine derivatives,as shown in Scheme 1. Details of reaction conditions and characterization data of compound 28 are summarized in the Supplementary data.
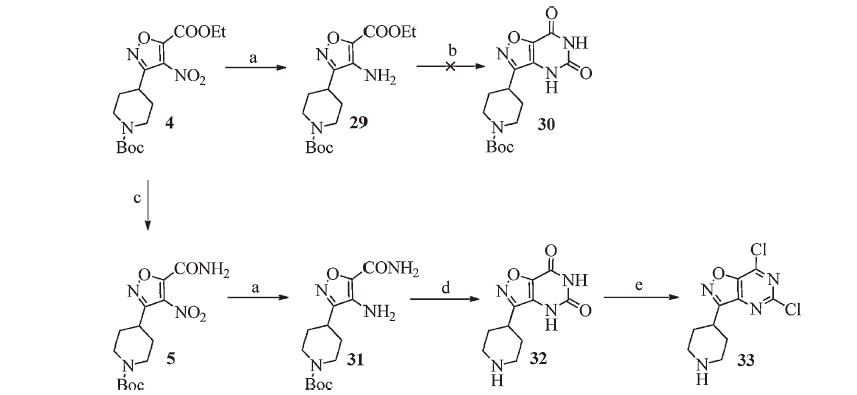
Under reflux,the reaction of compound 2 with hydroxylamine hydrochloride in EtOH did not yield 3,but the t-Boc deprotection product of 3. Most likely,the deprotection was due to the in situ generated hydrochloride during the condensation. When the reaction was carried out in the presence of NaHCO3,3 was obtained in 62% yield. We also found this reaction was highly temperature dependent. At 60 ℃ and 50 ℃,3 was isolated in 77% and 95% yield, respectively. Notably,intermediate 3 was an inseparable mixture of cis- and trans-isomers (cis/trans = 6/1 by 1H NMR) and used ‘‘as is’’ in the next step. In a one-pot procedure,3 was first condensed with 2-chloro-2-oxoacetate in the presence of TEA. Complete disappearance of 3 was observed by TLC after 24 h,suggesting both cis- and trans-3 reacted with 2-chloro-2-oxoacetate. Only the intermediate derived from the cis-isomer cyclized to furnish isoxazole 4. No inter-conversion between cis- and trans-isomers was observed at any stage of the reaction.
As shown in Scheme 4,intermediate 4 was reduced to 29. Direct cyclization of 29 with urea at high temperature failed to yield any appreciable amount of 30 due to charring. Intermediate 4 was converted to amide 5 by aminolysis using NH3 in methanol. Intermediate 5 was reduced to 31 in high yield (90%). Reaction of 31 with triphosgene gave 32 as its hydrochloride salt in high yield (85%). Treatment of 32 with POCl3 and N,N-dimethylaniline under reflux gave 33 cleanly,in which the N-Boc group was removed during the reaction (confirmed by LC-MS). Compound 33 exhibited excellent aqueous solubility and could not be isolated during workup. Based on these results,we decided to exchange the N-substituent on the piperidine from t-Boc- to benzyl (5 to7 via 6) as shown in Scheme 1. Details of reaction conditions and characterization data related to compounds 29,31,and 32 are summarized in the Supplementary data.

|
Download:
|
| Scheme. 4.Constructing the fused pyrimidine. Reagents and conditions: (a) Zn, NH4Cl, EtOH/water = 1/1, 0 ℃-r.t., 3 h, compound 29 (79% yield), compound 31 (90% yield). (b) Urea, 210 ℃. (c) NH3/MeOH, r.t., 3 h, 96%. (d) Triphosgene, anhydrous dioxane, reflux, 1 h, 85% (hydrochloride salt). (e) N,N-Dimethylaniline, POCl3, reflux, 8 h. | |
The transformation of 10 to 11 using Suzuki coupling was a problematic step.Weexamined the commonly used catalysts,such as Pd(PPh3)4,Pd(OAc)2,PdCl2(dppf),and Pd2(dba)3,but all failed to produce product 11. Pd(OAc)2 in combination with different phosphine ligands,such as 2-(di-tert-butylphosphino)biphenyl, butyldi-1-adamantylphosphine,and 2-(dicyclohexylphosphino)- biphenyl,also did not yield any desired product. The starting material was recovered in all cases. Shen [19] reported Pd(PCy3)2Cl2 was an effective catalyst for the activation of the aryl chlorides in the Suzuki reaction. He speculated that the electron-rich nature of PCy3 might enhance the oxidative insertion of palladium into the Ar-C1 bond and that the increased steric encumbrance of the ligand also might facilitate the dissociation of the ligands from the palladium complex. Using Pd(PCy3)2Cl2 as the catalyst in the presence of CsF at 100 ℃ for 48 h (Scheme 1),11 was obtained in 34% yield with 21% 10 recovered from the reaction mixture. Some isoxazole ring-opening product via the reductive cleavage of the N-O bond was also observed. Further optimization, such as prolonging reaction time,higher reaction temperature and various bases (K3PO4,K2CO3 and KF) did not improve the yield. Higher reaction temperature and/or longer reaction time all led to decreased yield due to greater extent of the isoxazole ring-opening reaction.
3.2. Biological evaluationCytotoxicity of compounds 11-23 against BT-474 cells was evaluated by CCK-8 assay. As summarized in Table 1,compared to other derivatives,11 showed lower toxicity to BT-474 cells, indicating the urea moiety on the phenyl ring was required for activity. While keeping R2 constant (R2 = benzyl),various substituent on the urea (R1) were examined. With large aromatic groups, 12-14 showed substantial cytotoxicities,with IC50s in the low to mid single digit micromolar range. In comparison,most alkyl derivatives,including the unsubstituted derivative 15,showed greater potency,with IC50s in the low single digit micromolar range,except the branched alkyl (17,R1 = -CH(CH3)2, IC50 = 8.148 μmol/L). While keeping R1 = CH3,we also briefly investigated the effects of N-substituents on the piperidine ring. N-ethyl substitution (23) and unsubstituted piperidine (22) all yield potent inhibitors,with IC50s in the low single digit micromolar range,indicating this site is amendable for further modifications.
| Table 1The cytotoxicity of compounds 11–23 in BT-474 cells. |
In combination with other data in hand,compounds 20 and 21 were further tested in biochemical assay to confirm their inhibition against PI3Kδ. As shown in Table 2,both 20 and 21 are potent PI3Kδ inhibitors with IC50s values of 0.286 μmol/L and 0.452 μmol/L, respectively. In the same assay,CAL-101 showed an IC50 of 0.036 μmol/L [24]. These data indicate their anti-proliferative activities are most likely due to their inhibition of the PI3Kδ kinase.
| Table 2The inhibitory activities of compounds 20 and 21 against PI3Kδ. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In summary,an efficient syntheses of novel 3-(piperidin-4- yl)isoxazolo[4, 5, d]pyrimidine scaffold was designed and developed. Using this method,a series of isoxazolo[4, 5, d]pyrimidine derivatives has been prepared. Their cytotoxicity to BT-474 cells was evaluated by CCK-8 assay. Most of the derivatives displayed inhibitory potency against the proliferation of BT-474 cells. Preliminary SAR information from these compounds can be used to guide further exploration. Although the lead compounds were also shown to be potent inhibitors of PI3Kδ kinase,indicating their anti-cancer activity could be through PI3K pathway,further studies are needed to optimize their activities and to confirm their mechanism of action.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 81172914) and Tang Aoqing Professorship research grant from Jilin University,China and Changchun Discovery Sciences,Ltd. The authors wish to thank Prof. Xu Bai for insightful discussions on chemical synthesis.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version,at http://dx.doi.org/10.1016/j.cclet.2015. 05.041.
| [1] | R. Williams, A. Berndt, S. Miller, W.C. Hon, X.X. Zhang, Form and flexibility in phosphoinositide 3-kinases, Biochem. Soc. Trans. 37(2009) 615-626. |
| [2] | P.X. Liu, H.X. Cheng, T.M. Roberts, J.J. Zhao, Targeting the phosphoinositide 3-kinase pathway in cancer, Nat. Rev. Drug Discov. 8(2009) 627-644. |
| [3] | L.C. Cantley, The phosphoinositide 3-kinase pathway, Science 296(2002) 1655-1657. |
| [4] | E. Ciraolo, F. Morello, E. Hirsch, Present and future of PI3K pathway inhibition in cancer:perspectives and limitations, Curr. Med. Chem. 18(2011) 2674-2685. |
| [5] | B.H. Jiang, L.Z. Liu, PI3K/PTEN signaling in angiogenesis and tumorigenesis, Adv. Cancer Res. 102(2009) 19-65. |
| [6] | B. Markman, R. Dienstmann, J. Tabernero, Targeting the PI3K/Akt/mTOR pathwaybeyond rapalogs, Oncotarget 1(2010) 530-543. |
| [7] | R. Marone, V. Cmiljanovic, B. Giese, M.P. Wymann, Targeting phosphoinositide 3-kinase-moving towards therapy, Biochim. Biophys. Acta 1784(2008) 159-185. |
| [8] | A. Carnero, Novel inhibitors of the PI3K family, Expert Opin. Investig. Drugs 18(2009) 1265-1277. |
| [9] | C.M. Dehnhardt, A.M. Venkatesan, E.D. Santos, et al., Lead optimization of N-3-substituted 7-morpholinotriazolopyrimidines as dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitors:discovery of PKI-402, J. Med. Chem. 53(2010) 798-810. |
| [10] | A.J. Folkes, K. Ahmadi, W.K. Alderton, et al., The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[32-d] pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer, J. Med. Chem. 51(2008) 5522-5532. |
| [11] | M.T. Burger, S. Pecchi, A. Wagman, et al., Identification of NVP-BKM120 as a potent, selective, orally bioavailable class I PI3 kinase inhibitor for treating cancer, ACS Med. Chem. Lett. 2(2011) 774-779. |
| [12] | J.J. Hale, C.L. Lynch, C.G. Caldwell, et al., Pyrrolidine modulators of CCR5 chemokine receptor activity, US/2002/0094989. |
| [13] | J.A. Deceuninck, D.K. Buffel, G.J. Hoornaert, A pathway to 3-(β-D-ribofuranosyl)-4-nitro-5-ethoxycarbonyl-isoxazoles, useful in the synthesis of pyrazofurin analogues, Tetrahedron Lett. 21(1980) 3613-3616. |
| [14] | H. Liu, X.H. He, H.S. Choi, et al., Compounds and compositions as inhibitors of cannabinoid receptor 1 activity, WO/2006/047516. |
| [15] | U. Niewohner, H. Haning, T. Lampe, et al., Isoxazolo pyrimidinones and the use thereof, US/2003/149033. |
| [16] | M.P. Prasad, B. Laxminarayan, Compositions, synthesis, and methodes of using indanone based cholinesterase inhibitors, WO/2008/073452. |
| [17] | V.J. Cunera, Z. Arie, A.K. Semiramis, et al., Ureidoaryl- and carbamoylaryl-morpholino-pyrimidine compounds, their use as mTOR kinase and PI3 kinase inhibitors, and their synthesis, WO/2010/120994. |
| [18] | T.P. Heffron, B.Q. Wei, A. Olivero, et al., Rational design of phosphoinositide 3-kinase α inhibitors that exhibit selectivity over the phosphoinositide 3-kinase β isoform, J. Med. Chem. 54(2011) 7815-7833. |
| [19] | W. Shen, Palladium catalyzed coupling of aryl chlorides with arylboronic acids, Tetrahedron Lett. 38(1997) 5575-5578. |
| [20] | A.M. Venkatesan, C.M. Dehnhardt, E.D. Santos, et al., Bis (morpholino-13, 5-triazine) derivatives:potent adenosine 50-triphosphate competitive phosphatidylinositol-3-kinase/mammalian target of rapamycin inhibitors:discovery of compound 26(PKI-587), a highly efficacious dual inhibitor, J. Med. Chem. 53(2010) 2636-2645. |
| [21] | Q.Z. Zheng, K. Cheng, X.M. Zhang, et al., Synthesis of some N-alkyl substituted urea derivatives as antibacterial and antifungal agents, Eur. J. Med. Chem. 45(2010) 3207-3212. |
| [22] | B.V. Yang, D. O'Rourke, J.C. Li, Mild and selective debenzylation of tertiary amines using a-chloroethyl chloroformate, Synlett 3(1994) 195-196. |
| [23] | F. Pettersson, P. Svensson, S. Waters, N. Waters, C. Sonesson, Synthesis, pharmacological evaluation and QSAR modeling of mono-substituted 4-phenylpiperidines and 4-phenylpiperazines, Eur. J. Med. Chem. 62(2013) 241-255. |
| [24] | B.J. Lannutti, S.A. Meadows, S.E.M. Herman, et al., CAL-101, a p110δ selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability, Blood 117(2011) 591-594. |