 2015, Vol.26
2015, Vol.26 

b Department of Chemistry and Centre for Atomic Engineering of Advanced Materials, Anhui University, Hefei 230601, China
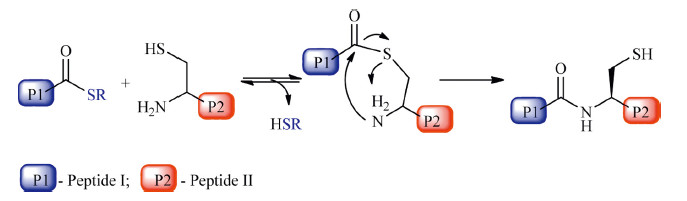
Native chemical ligation (NCL) was reported by Kent et al. in 1994 [1]. NCL corresponds to the formation of a polypeptide via the ligation of an unprotected peptide-a-thioester and an N-terminal cysteine peptide (Scheme 1). So far NCL has been widely used in chemical synthesis of proteins and peptides [2, 3, 4, 5, 6, 7, 8, 9]. Mechanistic understandings on NCL reaction will benefit the development of more powerful strategies [10, 11, 12, 13, 14, 15, 16, 17, 18]. The mechanism of NCL reaction mainly consists of three steps: the thioesterification between the N-terminal Cys and C-terminal thioester,the transthioesterification on the formed thioester intermediate,and the final intramolecular S→N acyl transfer step (Scheme 2) [10, 11, 12, 13, 14, 15]. With the aid of Density Functional Theory (DFT) calculation methods,we recently confirmed that the thiol-thioester exchange process is the rate determining step for thiol catalyzed NCL reactions [16, 17]. To clarify the key structural parameters in NCL reaction,in the present study we carried out DFT calculations on the thiol-thioester exchange step between PhS- and several thioesters.

|
Download:
|
| Scheme. 1.Illustrative figure of NCL reaction. | |

|
Download:
|
| Scheme. 2.The proposed mechanism of thiol catalyzed NCL reaction. | |
Recently,DFT calculations have beenwidely used inmechanistic studies of organic and bio-organic reactions [18, 19]. In this paper, M06-2X/6-31G(d) [20, 21, 22] method was used for the gas-phase optimizations of all species. Frequency calculationswere performed with the same method to verify the concerned compound to be minimum(with zero imaginary frequency) or transition state (with one imaginary frequency) and gain the thermal corrections to Gibbs free energy. For each transition state,IRC (intrinsic reaction coordinate) calculations were carried out to ascertain the correct connection between the transition state and the concerned reactant and product. The gas phase energies (total electronic energywiththe thermal correction to Gibbs free energy) were used for all discussions unless otherwise noted. The solvent effect has also been taken into account by performing single point energy calculations (the details are given in the Supporting information). All these calculations were performed with Gaussian09 software [23].
3. Results and discussionSix thioesters (Fig. 1) were studied and the thiol-thioester exchange step (between each of them and thiophenol anion) was examined. For clarity reasons,the transition state in the thiol- thioester exchange step and the product related to each Cn are named as TS-Cn and P-Cn,respectively.

|
Download:
|
| Fig. 1.The selected thioester substrates. | |
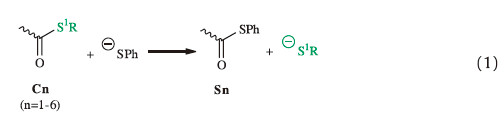
We used C1 as an example to perform detailed analysis on the structural and energetic changes in the thiol-thioester exchange step (Fig. 2). From C1,the approach of the thiophenol anion (PhS-) results in the automatic lengthening of the C-S1 bond,and in TS-C1 the C-S1 and C-S2 bond distances are 1.991 and 2.137Å . Thereafter, the formation of the C-S2 bond compensates the energetic necessity of the C-S1 bond dissociation,and thus the energy becomes lower until the formation of the subsequent intermediate P-C1. In P-C1,the C-S1 and C-S2 bond distances are 2.056 and 2.030Å,respectively. The C-S2 bond distance in the separated PhS2-connected thioester S1 (Eq. 1 and Table 1) is only 1.810Å. Therefore,P-C1 retained the weak interactions between the thioester moiety and the leaving S1R group (R = Et for C1). This effect weakens the interaction between the carbonyl group and the PhS2- group,and results in the relatively longer C-S2 bond distance in P-C1 (than that in S1). The transformation of C1→P-C1 is endergonic by 18.8 kcal/mol,and the activation barrier of this step is 19.8 kcal/mol (Fig. 2). Therefore,the transition state is a latetransition state. TS-C1 is energetically and structurally close to the formed intermediate P-C1.

|
Download:
|
| Fig. 2.The relative Gibbs free energy and the optimized structures of species involved in the thiol–thioester exchange step from C1. The bond distances are given in angstrom. | |
| Table 1 The relative free energies, and the C-Sx bond distances of all species in the thiol–thioester exchange steps on Cn (n = 1–6). |
The calculation results for the other thioesters gave similar conclusions. The thiol-thioester exchange process of each thioester is endergonic,and the C-S2 bond formation occurs with a simultaneous C-S1 bond cleavage. All the transition states (TS-Cn) are late transition states,and they are structurally and energetically close to the product (P-Cn). The C-S1 and C-S2 bond distances in P-Cn are about 2.0Å,indicating that C-S1 is partially dissociated while C-S2 is partially formed in P-Cn (the C-S2 in all separated product Sn are all about 1.8Å). Accordingly,in all the concerned products,the leaving S1R group remains weakly coordinated to the carbonyl group after the thiol-thioester exchange step.
The activation barrier of different transition states varies a lot (from 3.8 kcal/mol on C6 to 23.6 kcal/mol on C4). The relative free energy barriers of the aryl thioesters (C5 and C6,< 10 kcal/mol) are significantly lower than those of the alkyl thioesters (C1-C4, >15.0 kcal/mol),while the high energy barrier of C4 might be originated from the steric hindrance. It is expected that the thiol- thioester exchange step will be limited by steric hindrance (disfavors the approaching of the thiol group),and therefore we examines the electronic effect of different thioesters below.
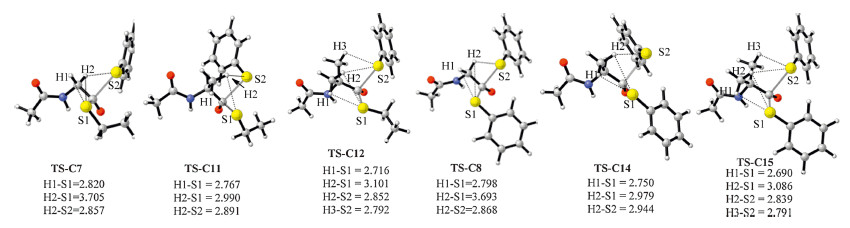
The ethyl thioester C7 and the benzene thioester C8 were chosen as samples to compare the electronic effect of alkyl and aromatic substituents on thioester. The substituent effect of the aromatic group on the thiol group was also examined for the comparison of the detailed electronic effect on the aromatic thiol group (C9 and C10 in Fig. 3). The free energy barriers of the thiol- thioester exchange step on C7 (13.3 kcal/mol,Table 2) are higher than those on C8-C10 (10.9,11.4,and 10.3 kcal/mol). The reason is related to the relatively stronger C-S(alkyl) bond than the C-S(aromatic) bond. This proposal is supported by the calculation results in Table 2: the C-S1 bond distance in C7 is shorter than the related ones in C8-C10,while the C-S1 bond dissociation enthalpy (BDE) of C7 is slightly higher than those of C8-C10. The differences of the free energy barriers between C7 and C8/C9/C10 (~2 kcal/mol) are significantly lower than those between C1/C2/ C3/C4 and C5/C6 (>8 kcal/mol). Therefore,although the aromatic thiol is a better leaving group,this electronic effect does not significantly influence the facility of the thiol-thioester exchange step. This conclusion also explains the high similarity between the energy barriers on C8-C10.

|
Download:
|
| Fig. 3.Selected modeling thioesters for the examination of the electronic effect on the thiol part. | |
| Table 2 The calculation results on the thiol–thioester exchange step on Cn (n = 7–10). |
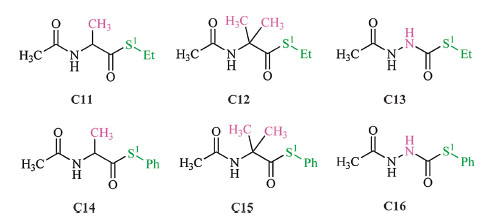
As the electronic effect of the thiol group is insignificant,we next examined the electronic effect of the carbonyl group. In accordance with the structures of thioesters in Fig. 1,the modeling thioesters of C11-C16 (Fig. 4) are used for these discussions,and the related calculation results are given in Table 3. First,the relative barriers of the a-NH substituted thioesters are significantly higher than the related ones of the a-CH2 substituted thioesters (C13 vs. C11/C12; C16 vs. C14/C15). Second,the activation barriers of the alkyl (on thiol group) thioesters are higher than the related aromatic thioesters (C11 vs. C14; C12 vs. C15; C13 vs. C16). Third,the substitution of the methyl group (s) on the a-C atom of C7,C11 and C12 facilitates the thiol-thioester exchange step,and the barriers of the aromatic thioesters (C8,C14 and C15) also follow the same trend.

|
Download:
|
| Fig. 4.Selected modeling thioesters for the examination of the substituent effect on the carbonyl group. | |
| Table 3 The calculation results on the thiol–thioester exchange step on C11-C16. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Efforts were made to understand the above observations. First, according to our recent studies the n→π* interaction of the proline between the aNatom and the carbonyl group increases the LUMO orbital energy of peptidyl-prolyl-thioester,and thus results in the relatively higher energy barriers of the thiol-thioester step. This conjugation effect also explains the relatively higher energy barriers of the thiol-thioester exchange steps on C13 and C16 (relative to the other thioesters in Table 3). Second,the above discussion implies the relatively stronger C-S bond strengths in the alkyl thioesters than the aromatic thioesters,and thus explains the relatively higher energy barriers of the former cases. Third, analyzing the optimized structures of TS-C7 and TS-C11/TS-C12, we found that the stronger hyper-conjugation interactions in the latter cases are mainly responsible for the relatively lower energy barriers. We take the comparison between TS-C7 and TS-C11 as an example. In TS-C7,two hyper-conjugation effects exist between the α-C-H bond and the -S1Et/-S2Ph group,and the related bond distances are both around 2.82Å(Fig. 5). By contrast,three hyperconjugation interactions are present in TS-C11,while the shortest H(α-methyl)-S(thiol) bond distance is 2.767Å. Note that the proposed hyper-conjugation effect (between the C-Hs bond of the substituted methyl group and the p orbitals of the thiol groups) can also be seen in the frontier orbital analysis of TS-C11 (Fig. 6). The same reason explains the relative lower free energy barrier of TS-C12 than TS-C11,and that TS-C15 < TS-C14 < TS-C8. Similarly, the relatively longer H(1)-S(1) bond distance in the alkyl thiol group involved transition states TS-C7/TS-C11/TS-C12 than the related aryl ones in TS-C8/TS-C14/TS-C15 indicates the weaker hyper-conjugation interactions in the former cases,and thus explains the higher barriers therein.

|
Download:
|
| Fig. 5.The optimized structures of TS-C7, TS-C8, TS-C11, TS-C12, TS-C14 and TS-C15. The bond distances are given in angstrom. | |
{kind=link}

|
Download:
|
| Fig. 6.The frontier orbitals of TS-C11. | |
{kind=link}
We noticed that the high steric hindrance significantly disfavors the thiol-thioester exchange step. When the steric hindrance is insignificant,the electronic effect becomes important in determining the facility of the thiol-thioester exchange step. That is,the aromatic thiol group is a relatively better leaving group than the alkyl thiol group. The conjugations between the N atom (on the side chain of the carbonyl group) and the carbonyl group disfavors the thiol-thioester exchange step,while the hyper-conjugation between the α-C substituted alkyl groups and the thiol groups favors the exchange step.
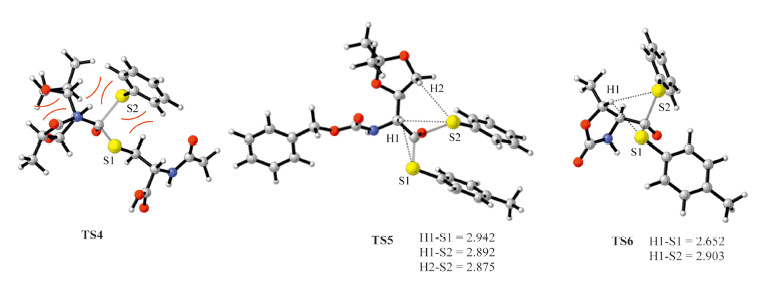
Finally,we made effort to understand the distinct energy barriers on C1-C6. First,the highest activation barrier on C4 among all the examined ones is caused by the high steric hindrance between the four bulky substituents around the carbonyl group (Fig. 7). Second,except for the high steric hindrance,the significantly lower energy barriers on C5 and C6 could be attributed to two folds: the aromatic thiol groups are better leaving groups than alkyl thiol groups,and the alkyl substituents on the α-C atoms leads to the stronger hyperconjugations with the thiol groups.Meanwhile,the slightly lower energy barrier of TS-C6 (relative to TS-C5) is caused by the slightly stronger hyper-conjugations (the H1-S1 bond distance in TS-C6 is significantly shorter than all the H-S bond distances in TS-C5,Fig. 7). Finally,the relatively higher energy barrier of TS-C3 (than TS-C1 and TS-C2) can be understood from the involvement of the -NH group in TS-C3. The relative barrier of TS-C2 is lower than that of TS-C1,presumably because of the conjugation between the benzene,amide and the thioester groups in C2. This effect makes the carbon center on C2 less electron deficient and more labile to dissociate the anionic -SEt group in the thiol-thioester exchange step.

|
Download:
|
| Fig. 7.The optimized structures of TS-C4, TS-C5 and TS-C6. | |
{kind=link}
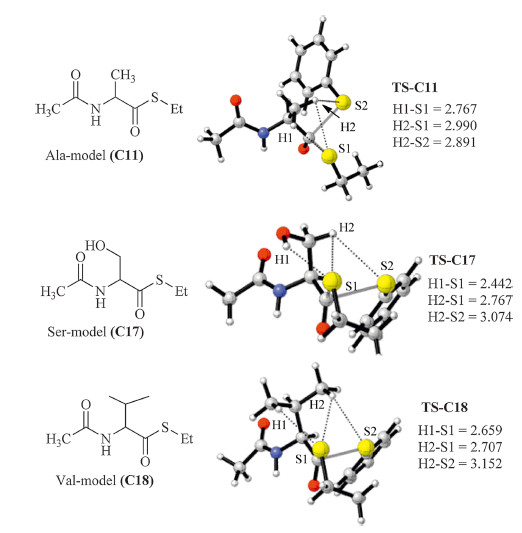
Some experimental observations in NCL reaction can also be understood from the aforementioned conclusions [24, 25, 26, 27, 28, 29]. For instance,according to the recent studies on the relative reaction rates of different thioesters (LYRAX-thioester (X = amino acid)) and CRANK,the NCL rates of Val (X = Val) was significantly lower than that of the Ala. The results are understandable because the steric hindrance of the side chain of Val and the thiol groups are significantly higher [29]. Indeed,the calculation results indicate that the activation barrier of the thiol-thioester step of the Val thioester (C17) is significantly higher than the Ala thioesters (C11, the energy barriers are 20.1 and 11.2 kcal/mol,respectively) (Fig. 8). On the other hand,the N-to-carbonyl conjugation also explains the relatively lower NCL activity of proline thioesters (relative to the alanyl thioester) recently reported by Kent et al. [26].

|
Download:
|
| Fig. 8.The modeling complexes and the related structures of TS-C11, TS-C17 and TS-C18. | |
{kind=link}
Density function theory calculations have been performed on the rate-determining step of the NCL reaction. The calculation results indicate that the thiol-thioester exchange step on all the concerned thioesters occur via the concerted C-S(Ph) bond formation and the C-S(R) bond (on the thioester substrate) dissociation step. The high steric hindrance is the main reason for the high energy barriers of the thiol-thioester exchange step. Meanwhile,the electronic effect becomes important as long as the steric hindrance is insignificant: the aryl thiol group is a relatively better leaving group than the alkyl thiol group; the conjugation between the side chain and the carbonyl group of thioester disfavors the thiol-thioester exchange step,while the hyperconjugation effect between the side chains of the carbonyl group and the leaving and approaching thiol groups facilitates the concerned thiol-thioester exchange step.
AcknowledgmentsWe appreciate NSFC (No. 21202006) and FRFCU (No. FRF-TP- 14-015A2) for financial supports and Super-computer Center of Shanghai and Shenzhen for technical supports.
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.07.003.
| [1] | P.E. Dawson, T.W. Muir, L.C. Lewis, S.B.H. Kent, Synthesis of proteins by native chemical ligation, Science 266(1994) 776-779. |
| [2] | P. Thapa, R.Y. Zhang, V. Menon, et al., Native chemical ligation:a boon to peptide chemistry, Molecules 19(2014) 14461-14483. |
| [3] | S. Stanchev, Z. Zawada, L. Moninc ová, et al., Synthesis of lucifensin by native chemical ligation and characteristics of its isomer having different disulfide bridge pattern, J. Pept. Sci. 20(2014) 725-735. |
| [4] | T. Nakamura, A. Shigenaga, K. Sato, et al., Examination of native chemical ligation using peptidyl prolyl thioesters, Chem. Commun. 50(2014) 58-60. |
| [5] | H. Kawashima, T. Kuruma, M. Yamashita, et al., Synthesis of an O-acyl isopeptide by using native chemical ligation in an aqueous solvent system, J. Pept. Sci. 20(2014) 361-365. |
| [6] | J.S. Zheng, S. Tang, Y.K. Qi, et al., Chemical synthesis of proteins using peptide hydrazides as thioester surrogates, Nat. Protoc. 8(2013) 2483. |
| [7] | H. van de Langemheen, M. van Hoeke, H.C. Quarles van Ufford, et al., Scaffolded multiple cyclic peptide libraries for protein mimics by native chemical ligation, Org. Biomol. Chem. 12(2014) 4471-4478. |
| [8] | Y.M. Li, Y.T. Li, M. Pan, et al., Irreversible site-specific hydrazinolysis of proteins by use of sortase, Angew. Chem. Int. Ed. 53(2014) 2198-2202. |
| [9] | C.T.T. Wong, C.L. Tung, X.C. Li, Synthetic cysteine surrogates used in native chemical ligation, Mol. BioSyst. 9(2013) 826-833. |
| [10] | Q.Q. He, G.M. Fang, L. Liu, Design of thiol-containing amino acids for native chemical ligation at non-Cys sites, Chin. Chem. Lett. 24(2013) 265-269. |
| [11] | L.R. Malins, N.J. Mitchell, R.J. Payne, Peptide ligation chemistry at selenol amino acids, J. Pept. Sci. 20(2014) 64-77. |
| [12] | R.E. Thompson, X.Y. Liu, N. Alonso-García, et al., Trifluoroethanethiol:an additive for efficient one-pot peptide ligation-desulfurization chemistry, J. Am. Chem. 136(2014) 8161. |
| [13] | J.S. Zheng, H.N. Chang, F.L. Wang, L. Liu, Fmoc synthesis of peptide thioesters without post-chain-assembly manipulation, J. Am. Chem. Soc. 133(2011) 11080. |
| [14] | L.E. Canne, S.J. Bark, S.B.H. Kent, Extending the applicability of native chemical ligation, J. Am. Chem. Soc. 118(1996) 5891-5896. |
| [15] | P.E. Dawson, M.J. Churchill, M.R. Ghadiri, S.B.H. Kent, Modulation of reactivity in native chemical ligation through the use of thiol additives, J. Am. Chem. Soc. 119(1997) 4325-4329. |
| [16] | C. Wang, Q.X. Guo, Y. Fu, Theoretical analysis of the detailed mechanism of native chemical ligation reactions, Chem. Asian J. 6(2011) 1241-1251. |
| [17] | Q. Zhang, H.Z. Yu, J. Shi, Orbital interactions in native chemical ligation reaction of proline thioesters, Acta. Phys. Chim. Sin. 29(2013) 2321-2331. |
| [18] | D.H. Yu, J.N. Shao, R.X. He, M. Li, Mechanism of trifluoromethylation reactions with well-defined NHC copper trifluoromethyl complexes and iodobenzene:a computational exploration, Chin Chem. Lett. 26(2015) 564. |
| [19] | X.N. Ke, C.M. Schienebeck, C.C. Zhou, X.F. Xu, W.P. Tang, Mechanism and reactivity of rhodium-catalyzed intermolecular[5+1] cycloaddition of 3-acyloxy-1,4-enyne (ACE) and CO:a computational study, Chin. Chem. Lett. 26(2015) 730. |
| [20] | E.C.B. Johnson, S.B.H. Kent, Insights into the mechanism and catalysis of the native chemical ligation reaction, J Am. Chem. Soc. 128(2006) 6640-6646. |
| [21] | H.Z. Yu, F. Fu, L. Zhang, et al., Accurate predictions of C-SO2R bond dissociation enthalpies using density functional theory methods, Phys. Chem. Chem. Phys. 16(2014) 20964-20970. |
| [22] | H.Z. Yu, Y.M. Yang, L. Zhang, Z.M. Dang, G.H. Hu, Quantum-chemical predictions of pKa's of thiols in DMSO, J. Phys. Chem. A 118(2014) 606-622. |
| [23] | M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2013. |
| [24] | T.M. Hackeng, J.H. Griffin, P.E. Dawson, Protein synthesis by native chemical ligation:expanded scope by using straightforward methodology, Proc. Natl. Acad. Sci. U. S. A. 96(1999) 10068-10073. |
| [25] | J.X. Wang, G.M. Fang, Y. He, et al., Peptide o-aminoanilides as crypto-thioesters for protein chemical synthesis, Angew. Chem. Int. Ed. 54(2015) 2194. |
| [26] | S.B. Pollock, S.B.H. Kent, An investigation into the origin of the dramatically reduced reactivity of peptide-prolyl-thioesters in native chemical ligation, Chem. Commun. 47(2011) 2342-2344. |
| [27] | C.Z. Sun, G. Luo, S. Neravetla, S.S. Ghosh, B. Forood, Native chemical ligation derived method for recombinant peptide/protein C-terminal amidation, Bioorg. Med. Chem. Lett. 23(2013) 5203-5208. |
| [28] | J.S. Zheng, S. Tang, Y.C. Huang, L. Liu, Development of new thioester equivalents for protein chemical synthesis, Acc. Chem. Res. 46(2013) 2475. |
| [29] | T. Küh, M. Chen, K. Teichmann, A. Stark, D. Imhof, Ionic liquid 1-ethyl-3-methylimidazolium acetate:an attractive solvent for native chemical ligation of peptides, Tetrahedron Lett. 55(2014) 3658-3662. |