 2015, Vol.26
2015, Vol.26 



b School of Pharmacy, Fudan University, Shanghai 200433, China
Natural products from ocean serve as a rich source of bioactive substances[1],for instance,alkaloids[2],cyclic peptides[3]and cyclic depsipeptides[4].Marine fungi and cyanobacteria,predominantly with structural skeletons as modified peptides,depsipeptides,polyketides and peptide-polyketide hybrids,have emerged as a valuable source of marine natural products with remarkable biological activities[5, 6].As a class of promising compounds in drug discovery,most of these secondary metabolites display a variety of physiological activities,including antimicrobial,antimalarial,cytotoxic and neorotoxic properties[7].Although a few of them were investigated for the application in potential cancer therapeutics[4, 8],most secondary metabolites are not well studied,thus require further research on either their structural modifications or modes of actions[9].
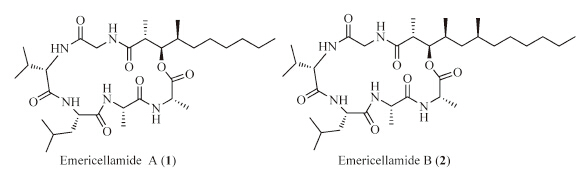
Emericellamides A and B (Fig.1) are two secondary metabolites of marine cyclic depsipeptides,isolated from the co-culture of the marine-derived fungus Emericella sp.and actinomycete Salinispora arenicola by Fenical and his co-workers[10].Their structures were determined based on chemical and spectroscopic methods,and the absolute configurations were assigned by Marfey's method[11]and the modified Mosher method[12].Emericellamides A and B exhibit antimicrobial activity against methicillin-resistant Staphylococcus aureus (MIC 3.8 mmol/L and 6.0 mmol/L,respectively),as well as cytotoxicity against HCT-116 human colon carcinoma cell line (IC50 23 mmol/L and 40 mmol/L,respectively).The biosynthetic pathway for emericellamides was identified in 2008[13].

|
Download:
|
| Fig. 1.The structures of emericellamides A (1) and B (2). | |
{kind=link}
Due to the attractive bioactivities and intriguing structure and their analogous,considerable efforts have been contributed to their synthesis,and several enantioselective synthetic methods were reported[14, 15, 16, 17, 18].As a continuation of our interest in pursuing diverse synthesis of alkaloids[19, 20],and investigating their structure-activity relationship,we synthesized several secondary metabolites[16, 21, 22]including emericellamide A[16].Herein we present a general and efficient method for total synthesis of emericellamide B.
2.ExperimentalTHF was distilled from sodium/benzophenone.Reactions were monitored by thin layer chromatography (TLC) on glass plates coated with silica gel with fluorescent indicator.Flash chromatography was performed on silica gel (300-400) with petroleum ether/EtOAc as eluent.Optical rotations were measured on a polarimeter with a sodium lamp.HRMS were measured on a LCMSIT-TOF apparatus.IR spectra were recorded using film on a Fourier Transform Infrared Spectrometer.NMR spectra were recorded at 400 MHz or 500 MHz,and chemical shifts are reported in δ (ppm) referenced to an internal TMS standard for 1H NMR and CDCl3(77.0 ppm) for 13C NMR.1H NMR and 13C NMR spectra of compounds 5-10,12-16,18 and emericellamide B are put in Supporting information.
2.1.(4S,5R,6S,2E)-Ethyl-7-benzyloxy-5-(tert-butyldimethylsilyloxy)-4,6-dimethylhept-2-enoate (5)To a suspension of NaH (1.16 g,31.37 mmol) in dry THF (60 mL) was added dropwise a solution of triethyl phosphonoacetate (6.6 mL,32.80 mmol) at 0 ℃ under argon atmosphere.After being stirred for 30min,the mixture was cooled to-78 ℃ and a solution of aldehyde 4(5.0 g,14.26mmol) in THF (10 mL) was added drop wise.The resulting mixture was stirred for 1 h at 0 ℃,then a phosphate buffer solution (20 mL) and Et2O (100mL) were added.The mixture was allowed to warm to room temperature,and the organic layer was separated,washed with saturated NH4Cl solution and brine,dried over anhydrous Na2SO4,filtered and concentrated.The residue was purified by chromatography on silica gel to give 5(5.78 g,yield 98%) as a colorless oil.[α]D25 -28.6(c 0.44,CHCl3);IR (film,cm-1):υmax 2957,2929,1720,1651,1464,1366,1259;1H NMR (400 MHz,CDCl3):δ 7.36-7.27(m,5H),6.98(dd,1H,J=7.82,15.85 Hz),5.78(dd,1H,J=1.17,15.85 Hz),4.48(d,1H,J=12.13 Hz),4.45(d,1H,J=12.13 Hz),4.18(m,2H),3.62(dd,1H,J=4.69,5.29 Hz),3.50(dd,1H,J=5.29,9.39 Hz),3.29(dd,1H,J=7.43,9.00 Hz),2.58-2.51(m,1H),1.97(m,1H),1.28(t,3H,J=7.05 Hz),1.04(d,3H,J=6.65 Hz),0.98(d,3H,J=6.65 Hz),0.88(s,9H),0.03(s,3H);13CNMR (125 MHz,CDCl3):δ 166.7,152.8,138.6,128.3(2C),127.5(2C),127.4,120.5,72.9,72.2,60.1,40.3,38.1,26.0(3C),14.9,14.3,14.2,-3.9,-4.1;MS (ESI):443.2(M+Na+);HRMS (ESI) calcd.for (C24H40O4Si+Na+):443.2595,found:443.2629.
2.2.(4S,5R,6S)-Ethyl-7-benzyloxy-5-(tert-butyldimethylsilyloxy)-4,6-dimethylheptanoate (6)To a solution of compound 5(5.50 g,13.07 mmol) in MeOH (100 mL) and THF (30 mL) was added NiCl2·6H2O (1.56 g,6.54 mmol) at 0 ℃ under argon atmosphere,then NaBH4(1.00 g,26.15 mmol) was added in several portions.After being stirred for 1 h,the mixture was concentrated,filtered with celite,and washed with Et2O (100 mL).The organic phase was concentrated,and the residue was purified by chromatography on silica gel to give 6(5.39 g,yield 97%) as a colorless oil.[α]D25 -14.7(c 0.82,CHCl3);IR (film,cm-1):υmax 2957,2856,1736,1471,1254,1178,1093;1H NMR (400 MHz,CDCl3):δ 7.36-7.25(m,5H),4.50(d,1H,J=12.13 Hz),4.46(d,1H,J=12.13 Hz),4.12(m,2H),3.53(dd,2H,J=4.30,9.00 Hz),3.49(dd,1H,J=2.54,6.46 Hz),3.28(dd,1H,J=7.64,9.00 Hz),2.36-2.25(m,2H),1.98-1.92(m,1H),1.74-1.66(m,1H),1.53-1.47(m,2H),1.24(t,3H,J=7.04 Hz),0.95(d,3H,J=6.72 Hz),0.91-0.84(m,12H,CH3),0.02(s,3H),-0.52(s,3H);13C NMR (125 MHz,CDCl3):δ 173.9,138.7,128.3(2C),127.5(2C),127.3,76.8,73.0,72.9,60.2,38.0,35.8,32.7,29.9,26.1(3C),18.4,15.1,14.2,13.8,-3.8,
-4.1;MS (ESI):445.2(M+Na+);HRMS (ESI) calcd.for (C24H42O4Si+Na+):445.2752,found:445.2749.
To a solution of 6(5.20 g,12.36 mmol) in THF/H2O (150mL,v/v=3/1) was added LiOH·H2O (5.20 g,123.60 mmol) at room temperature.After being stirred for 6 h at 60 ℃,the mixture was quenched with 1 mol/L HCl and extracted with EtOAc (150 mL × 3).The combined organic layers were dried over anhydrous Na2SO4,filtered,and concentrated to yield 7(4.78 g,yield 98%),which was used in next step without further purification.[α]D25 -11.3(c 5.05,CHCl3);IR (film,cm-1):υmax 3030,2928,1709,1454,1383,1253,1093;1H NMR (400 MHz,CDCl3):δ 7.37-7.26(m,5H),4.51(d,1H,J=12.13 Hz),4.45(d,1H,J=12.13 Hz),3.55-3.49(m,1H),3.28(dd,1H,J=7.62,9.00 Hz),2.43-2.25(m,2H),1.99-1.92(m,1H),1.76-1.70(m,1H),1.66-1.57(m,1H),1.55-1.49(m,1H),0.96(d,3H,J=7.04 Hz),0.89-0.84(m,12H),0.03(s,3H),0.01(s,3H);13C NMR (125 MHz,CDCl3):δ 180.1,138.7,128.3(2C),127.6(2C),127.4,76.2,73.0,72.9,37.9,35.8,32.3,29.5,26.1(3C),18.4,15.1,13.9,-3.8,-4.1;MS (ESI):417.2(M+Na+);HRMS (ESI) calcd.for C22H38O4Si:394.2539(C22H38O4Si+Na+):417.2439,found 417.2459.
2.4.(R)-3-((4'S,5'R,6'S)-7'-Benzyloxy-5'-(tert-butyldimethylsilyloxy)-4',6'-dimethylheptanoyl)-4-benzyloxazolidin-2-one (8)To a solution of crude acid 7(5.30 g,13.43 mmol) and Et3N (3.7 mL,26.86 mmol) in dry THF (70 mL) was added pivaloyl chloride (1.95 mL,16.12 mmol) dropwise at -78 ℃ under argon atmosphere.After being stirred at this temperature for 2 h,oxazolidinone (2.40 g,13.70 mmol) and LiCl (1.70 g,40.29 mmol) were added in one portion,then the mixture was slowly warmed to room temperature and stirred overnight.The resulting mixture was diluted with water (100 mL) and ether (100 mL),the organic phase was separated and the aqueous layer was extracted with ether (100 mL × 3).The combined organic layers were washed with brine,dried and concentrated,and the residue was purified by chromatography on silica gel to give 8(6.44 g,yield 87%) as a colorless oil.[α]D25 -24.2(c 5.34,CHCl3);IR (film,cm-1):υmax 2956,2927,1784,1702,1384,1096;1H NMR (400 MHz,CDCl3):δ 7.36-7.20(m,10H),4.66(ddd,1H,J=3.32,6.12,13.10 Hz),4.51(d,1H,J=11.94 Hz),4.48(d,1H,J=11.94 Hz),4.21-4.13(m,2H),3.59-3.53(m,2H),3.33-3.28(m,2H),3.07-2.99(m,1H),2.89-2.81(m,1H),2.72(dd,1H,J=9.80,13.31 Hz),2.02-1.95(m,1H),1.81-1.73(m,1H),1.70-1.66(m,1H),1.63-1.55(m,1H),0.98(d,3H,J=6.85 Hz),0.92-0.87(m,12H,CH3),0.07(s,3H),0.04(s,3H);13C NMR (125 MHz,CDCl3):δ 173.3,153.4,138.8,135.3,129.4(2C),128.9(2C),128.3(2C),127.5(2C),127.4,127.3,76.8,73.0,72.9,66.1,55.1,38.0,37.9,
35.8,33.9,29.3,26.1(3C),18.4,15.1,13.9,-.3.6,-4.1;MS (ESI):554.3(M+H+);HRMS (ESI) calcd.for (C32H47NO5Si+Na+):576.3123,found:576.3152.
To a solution of amide 8(4.50 g,8.13 mmol) in THF (45 mL) was added a solution of NaHMDS (8.9 mL,1 mol/L in THF,8.94 mmol) dropwise at -78 ℃ under argon atmosphere,the mixture was stirred for 30 min,then MeI (1.50 mL,24.38 mmol) was added.After being stirred at this temperature for 3 h,the mixture was warmed to room temperature and stirred overnight.The reaction was quenched with H2O (50 mL) and extracted with Et2O (50 mL × 3).The combined organic extracts were washed with brine,dried and concentrated,and the residue was purified by chromatography on silica gel to give 9(3.40 g,yield 74%) as a colorless oil.[α]D25 -50.7(c 1.15,CHCl3);IR (film,cm-1):υmax 2928,1782,1698,1454,1385,1249,1208,1099;1H NMR (600 MHz,CDCl3):δ 7.33-7.17(m,10H),4.63-4.58(m,1H),4.48(d,1H,J=12.06 Hz),4.45(d,1H,J=12.06 Hz),4.11(d,2H,J=5.11 Hz),3.82(ddd,1H,J=4.74,7.02,11.52 Hz),3.55(dd,1H,J=4.32,9.00 Hz),3.45(dd,1H,J=2.82,6.48 Hz),3.26(dd,1H,J=8.04,9.00 Hz),3.21(dd,1H,J=3.30,13.36,Hz),2.73(dd,1H,J=9.60,13.36 Hz),1.95(ddd,1H,J=1.80,6.96,14.10 Hz),1.86(ddd,1H,J=3.96,10.18,14.05 Hz),1.57-1.53(m,1H),1.27(ddd,1H,J=4.62,9.72,14.05 Hz),1.20(d,3H,J=6.90 Hz),0.94(d,3H,J=6.90 Hz),0.86(s,9H),0.83(d,3H,J=6.72 Hz),0.01(s,3H),0.00(s,3H);13C NMR (150 MHz,CDCl3):δ 177.1,153.0,138.9,135.3,129.5(2C),128.9(2C),128.3(2C),127.5(2C),127.4(2C),77.9,73.1,73.0,66.0,55.3,39.1,38.2,
37.8,35.5,34.0,26.2(3C),19.0,18.5,15.1,13.9,-3.5,-4.0;MS (ESI):590.3(M+Na+);HRMS (ESI) calcd.for (C33H49NO5Si+Na+):590.3280,found:590.3305.
To a solution of amide 9(3.30 g,5.81 mmol) in dry THF (30 mL) was added a solution of LiBH4(8.7 mL,2.0 mol/L solution in THF,17.44 mmol) at 0 ℃ under argon atmosphere,then dry methanol (0.6 mL,17.44 mmol) was slowly added.After being stirred for 45 min at 0 ℃ and 1 h at room temperature,the mixture was cooled back to 0 ℃ and quenched carefully with a 1.0 mol/L aqueous solution of NaOH (30 mL).The mixture was extracted with Et2O (50 mL × 3),and the combined organic layers were washed with brine,dried and concentrated.The residue was purified by chromatography on silica gel to give alcohol 10(2.13 g,yield 93%) as a colorless oil.[α]D25 -11.9(c 3.0,CHCl3);IR (film,cm-1):υmax 3377,2957,2928,2856,1462,1381,1252,1069;1H NMR (400 MHz,CDCl3)δ 7.37-7.26(m,5H),4.50(d,1H,J=12.13 Hz),4.46(d,1H,J=12.13 Hz),3.56(dd,1H,J=5.09,9.00 Hz),3.50(dd,1H,J=4.11,10.47 Hz),3.44(dd,1H,J=3.32,5.67 Hz),3.36(dd,1H,J=6.65,10.47 Hz),3.25(dd,1H,J=7.04,9.00 Hz),2.03-1.96(m,1H),1.77-1.62(m,2H),1.49-1.36(m,2H),0.95(d,3H,J=6.85 Hz),0.93(d,3H,J=6.65 Hz),0.89-0.85(m,12H),0.03(s,3H),0.01(s,3H);13C NMR (125 MHz,CDCl3)δ 138.6,128.3(2C),127.6(2C),127.4,76.8,73.0,72.9,67.6,38.1,37.8,33.4,33.0,26.1(3C),18.5,17.8,15.5,15.3,-3.6,-4.0;MS (ESI):417.3(M+Na+);HRMS (ESI) calcd.for (C23H42O3Si+Na+):417.2803,found:417.2772.
2.7.(2S,3R,4S,6R,E)-1-Benzyloxy-3-(tert-butyldimethylsilyloxy)-2,4,6-trimethyldodec-7-en (12)To a solution of oxalyl chloride (0.94 mL,11.15 mmol) in dry CH2Cl2(30 mL) was added DMSO (1.60 mL,22.30 mmol) over 15 min at -78 ℃ under argon atmosphere.After being stirred for 30 min,a solution of the alcohol 10(2.20 g,5.57 mmol) in dry CH2Cl2(5.0 mL) was added slowly and the mixture was stirred for another 1 h,then Et3N (3.9 mL,27.85 mmol) was added drop wise and the mixture was allowed to warm to room temperature.The reaction was quenched with aqueous NaHSO4 solution (1 mol/L,30 mL),then the organic layer was separated,and the aqueous phase was extracted with CH2Cl2.The combined organic layerswere washed with saturated aqueous solution of NaHSO4,saturated aqueous NaHCO3,brine,and dried.The solvents were concentrated under reduced pressure to give aldehyde 11(2.0 g,yield 92%) as a colorless oil,which was used in next step without further purification.To a suspension of pentane-triphenylphosphonium bromine (11.17 g,27.02 mmol) in dry THF (50 mL) was added n-BuLi (16.8 mL,1.6 mol/L in hexane,26.8 mmol) drop wise at room temperature under argon atmosphere.After the red mixture was stirred for 30 min,a solution of the above crude aldehyde 11(2.10 g,5.35 mmol) in dry THF (5 mL) was added,and the resulting mixture was stirred for 2 h.The mixture was diluted with ethyl acetate and washed with saturated NaHCO3 aqueous,brine,dried over anhydrous Na2SO4.The solvents were concentrated,and the residue was purified by chromatography on silica gel to give 12(2.30 g,yield 97%) as a colorless oil.[α]D25 -5.07(c 4.48,CHCl3);IR (film,cm-1):υmax 2956,2856,1454,1379,1252;1H NMR (400 MHz,CDCl3)δ 7.35-7.25(m,5H),5.29(ddd,1H,J=7.24,11.15,14.47 Hz),4.99(dd,1H,J=9.97,11.15 Hz),4.50(d,1H,J=11.93 Hz),4.46(d,1H,J=11.93 Hz),3.55(dd,1H,J=4.30,8.90 Hz),3.40(dd,1H,J=3.13,6.06 Hz),3.26(dd,1H,J=7.63,8.43 Hz),2.53-2.46(m,1H),2.05-1.97(m,2H),1.97-1.89(m,1H),1.62-1.54(m,1H),1.36-1.27(m,5H),1.20(m,2H),0.96(d,3H,J=6.84 Hz),0.91-0.85(m,15H),0.82(d,3H,J=6.8 Hz),0.01(s,3H),0.00(s,3H);13C NMR (125 MHz,CDCl3)δ 138.8,135.9,128.9,128.3(2C),127.5(2C),127.3,78.6,73.1,72.9,42.7,37.7,34.2,32.2,29.3,27.3,26.1(3C),22.4(2C),18.4,
15.5,14.1,14.0,-3.6,-4.1;MS (ESI):469(M+Na+);HRMS (ESI) calcd.for (C28H50O2Si+Na+):469.3480,found:469.3464.
To a solution of 12(2.0 g,4.48 mmol) in MeOH (15 mL) was added a solution of HCl/MeOH (2 mol/L,5 mL) at room temperature,and the resulting mixture was stirred for 3 h.Then the mixture was concentrated in vacuo,the residue was diluted with water (20 mL) and Et2O (30 mL).The resulting mixture was separated,the aqueous phase was extracted with Et2O (30 mL × 3),and the combined organic layers were washed with saturated aqueous NaHCO3,brine,and dried over anhydrous Na2SO4.The solvents were concentrated,and the residue was purified by chromatography on silica gel to give 13(1.48 g,yield 100%) as a colorless oil.[α]D25+26.0(c 0.4,CHCl3);IR (film,cm-1):υmax 3507,2958,2926,2857,1455,1378,1095;1HNMR (600 MHz,CDCl3)δ 7.36-7.27(m,5H),5.30(ddd,1H,J=7.32,9.72,10.92 Hz),5.05(dd,1H,J=9.89,10.80 Hz),4.52(s,2H),3.60(dd,1H,J=4.14,8.94 Hz),3.48(dd,1H,J=8.22,8.94 Hz),3.38(d,1H,J=2.82 Hz),3.36-3.32(m,1H),2.57-2.51(m,1H),2.07-2.01(m,2H),1.98-1.91(m,1H),1.59-1.55(m,1H),1.38-1.29(m,5H),1.23(m,1H),0.94(d,3H,J=6.62 Hz),0.90(t,3H,J=7.12 Hz),0.85(d,3H,J=6.71 Hz),0.82(d,3H,J=7.03 Hz);13C NMR (150 MHz,CDCl3)δ 137.8,136.1,128.9,128.5(2C),127.8(2C),127.7,80.4,76.3,73.6,42.5,36.0,33.0,32.2,29.3,27.3,22.4,22.2,14.0,13.7,
12.0;MS (ESI):355.2(M+Na+);HRMS (MALDI/DHB) calcd.for (C22H36O2+Na+):355.2613,found:355.2609.
To a solution of alcohol 13(1.0 g,3.01 mmol),EDCI (2.91 g,152.0 mmol) in dry DCM (25 mL) was added N-Boc-Ala-OH (2.85 g,15.05 mmol) at 0 ℃ under argon atmosphere,and the mixture was stirred for 0.5 h,then DMAP (183 mg,1.50 mmol) was added in one portion.After being stirred for 3 h,the mixture was allowed to warm to room temperature and stirred for additional 6 h.The resulting mixture was diluted with CH2Cl2(30 mL),the organic layer was separated and the aqueous layer was extracted with CH2Cl2(30 mL × 2).The combined organic layers were washed with an aqueous solution of HCl (5%),saturated aqueous NaHCO3 solution and brine,dried and concentrated.The residue was purified by chromatography on silica gel to give 14(0.90 g,yield 60%;90% based on the recovery of starting material) as a light yellow oil.[α]D25 -11.3(c 0.56,CHCl3);IR (film,cm-1):υmax 3993,2962,2928,2871,1716,1497,1454,1366,1167;1H NMR (600 MHz,CDCl3)δ 7.34-7.27(m,5H),5.29(m;1H),5.06(d,1H,J=7.26 Hz),4.98(dd,1H,J=10.28,10.77 Hz),4.87-4.84(m,1H),4.45(s,2H),4.26(dd,1H,J=6.92,7.72 Hz),3.41(dd,1H,J=3.87,8.93 Hz),3.22(dd,1H,J=7.00,8.93 Hz),2.54-2.48(m,1H),2.07-1.94(m,2H),1.79-1.75(m,1H),1.46-1.42(m,9H),1.36(d,3H,J=7.14 Hz),1.39-1.25(m,4H),1.17(m,1H),1.06(m,1H),0.96(d,3H,J=6.84 Hz),0.92-0.88(m,6H),0.83(d,3H,J=6.72 Hz);13C NMR (150 MHz,CDCl3)δ 173.1,155.1,138.6,135.3,129.2,128.3(2C),127.7(2C),127.5,80.0,79.6,73.2,72.1,49.5,41.7,35.5,32.2,31.2,29.0,
28.4(3C),27.2,22.4(2C),22.0,14.4,14.0(2C);MS (ESI):526.4(M+Na+);HRMS (MALDI/DHB) calcd.for (C30H49NO5+Na+):526.3508,found:526.3507.
To a suspension of Pd/C (10%,80 mg) in CH3OH (10 mL) was added a solution of 14(800 mg,1.589 mmol) in CH3OH (2 mL).After the resulting mixture was stirred at room temperature under H2 atmosphere (1 atm) for 4 h,the catalyst was filtered off and the filtrate was concentrated in vacuo to give the crude alcohol,which was purified by chromatography on silica gel to give 15(658 mg,quantitative yield) as a light colorless oil.[α]D25 -28.5(c 0.85,CHCl3);IR (film,cm-1):υmax 3438,3368,2962,2927,1715,1682,1502,1456,1367,1215,1169,1068;1H NMR (600MHz,CDCl3)δ 5.01(d,1H,J=7.40 Hz),4.85(d,1H,J=10.26 Hz),4.30-4.24(m,1H),3.47-3.42(m,1H),3.46-3.43(m,1H),2.47(bs,1H),1.92-1.86(m,1H),1.86-1.81(m,1H),1.46-1.42(m,11H),1.40(d,3H,J=7.30Hz),1.30-1.16(m,9H),1.00(d,3H,J=6.96 Hz),0.90(d,3H,J=6.84 Hz),0.87(t,3H,J=7.10 Hz),0.80(d,3H,J=6.60Hz);13C NMR (150 MHz,CDCl3,25 ℃,TMS)δ 174.8,155.4,80.1,77.6,64.1,49.6,41.6,37.2,36.9,31.9,30.6,29.6,29.5,28.3(3C),26.9,22.7,19.8,18.4,14.1,14.0,13.6;MS (ESI):438.2(M+Na+);HRMS (MALDI/DHB) calcd.for (C23H45NO5+Na+):438.3195,found:438.3183.
2.11.(S)-((2'R,3'R,4'S,60S)-1'-(200-(Benzyloxy)-2"-oxoethylamino)-2',4',6'-tri-methyl-1'-oxododecan-30-yl)-2-(tertbutoxycarbonylamino) propanoate (16)To a stirred solution of 15(200 mg,0.482 mmol) in CCl4(5 mL) were added CH3CN (5 mL) and water (7.5 mL),then NaIO4(413 mg,1.93 mmol) and RuCl3·xH2O (5.0 mg,0.04 mmol) were added sequentially at room temperature.After being stirred for 4 h,the reaction was diluted with Et2O (20 mL) and stirred for 20 min to precipitate black RuO2.Then the mixture was dried and filtered through celite,the solid residue was washed with ether,and the combined organic phases were concentrated in vacuo to give the carboxylic acid (200 mg),which was used in next step without further purification.The above crude carboxylic acid (200 mg,0.466 mmol) and HOBt (69 mg,0.513 mmol) were added in dry DMF (4 mL) and stirred for 15 min at room temperature,then the reaction mixture was cooled to -15 ℃,EDCI (98 mg,0.513 mmol) was added in one portion and stirred for another 1.5 h at this temperature,H-Gly-OBn (85 mg,0.513 mmol) was added in one portion.After being stirred overnight,the mixture was quenched with H2O (10 mL),and extracted with Et2O (20 mL × 3).The combined organic layers were washed with water,saturatedaqueous NaHCO3 and brine,dried and concentrated.The residue was purified by chromatography on silica gel to give 16(238 mg,yield 88% over three steps) as a light colorless oil.[α]D25 -13.2(c 1.61,CHCl3);IR (film,cm-1):υmax 3325,2959,2927,1730,1717,1659,1525,1456,1366,1174;1H NMR (600 MHz,CDCl3)δ 7.37-7.31(m,5H),6.30(bs,1H),5.20-5.14(m,3H),5.06(m,1H),4.31-4.28(m,1H),4.06(d,2H,J=3.84 Hz),2.65(m,1H),1.93-1.87(m,1H),1.58-1.51(m,1H),1.42-1.39(m,10H),1.37(d,3H,J=7.20 Hz),1.30-1.18(m,9H),1.15(d,3H,J=7.01 Hz),1.04-0.98(m,1H),0.93-0.86(m,7H),0.83(d,3H,J=6.60 Hz);13C NMR (150 MHz,CDCl3)δ 173.7,172.6,170.1,155.2,135.2,128.6(2C),128.5(2C),128.3,79.7,78.0,67.2,49.4,43.6,41.4,41.1,36.5,31.9,31.4,29.7,
29.6,28.3(3C),26.7,22.7,20.1,18.4,14.6,14.1,13.9;MS (ESI):599.4(M+Na+);HRMS (MALDI/DHB) calcd.for (C32H52N2O7+Na+):599.3672,found:599.3659.
To a solution of 16(200 mg,0.347 mmol) in CH2Cl2(3 mL) was added TFA (3 mL) and stirred for 2 h at room temperature,then the solvent was evaporated in vacuo.The residue was dissolved in 1,2-dichloroethane (3 mL),the solvent was removed in vacuo and this process was repeated three times to give crude 17.HOBt (52 mg,0.347 mmol) was added to a solution of N-Cbz-Val-Leu-Ala-OH (137 mg,0.315 mmol) in dry DMF (5 mL) at -15 ℃.After 30 min of stirring,the resulting cold solution was treated with EDCI (73 mg,0.347 mmol) and stirred at -15 ℃ for 1.5 h,then NMM (76 mL,0.694 mmol) and a solution of amide 17 in DMF provided above were added before being warmed to room temperature overnight.The reaction was quenched with H2O (15 mL) and extracted with EtOAc (30 mL × 3).The combined organic extracts were washed with water,saturated aqueous NaHCO3 and brine,dried and concentrated.The residue was purified by chromatography on silica gel to give 18(201 mg,yield 65% over two steps) as a white solid.[α]D25 -34.7(c 0.36,CHCl3);IR (film,cm-1):υmax 3280,2958,2928,1741,1697,1634,1541,1454,1384,1210;1H NMR (600 MHz,DMSO-d6)δ 8.25(t,1H,J=5.80 Hz),8.01(d,1H,J=7.41 Hz),7.91-7.87(m,2H),7.38-7.28(m,11H),5.11(d,1H,J=13.14 Hz),5.09(d,1H,J=13.14 Hz),5.03(d,1H,J=13.08 Hz),5.00(d,1H,J=13.08 Hz),4.91(dd,1H,J=1.82,9.62 Hz),4.33-4.24(m,3H),3.89-3.82(m,2H),3.76(dd,1H,J=5.64,17.52 Hz),2.69-2.63(m,1H),1.97-1.91(m,1H),1.85-1.79(m,1H),1.63-1.57(m,1H),1.55-1.50(m,1H),1.44-1.40(m,2H),1.28-1.14(m,17H),1.02-0.97(m,1H),0.96(d,3H,J=7.02 Hz),0.87-0.77(m,21H);13C NMR (150 MHz,DMSO-d6)δ 173.9,171.9,171.5,171.4,170.3,156.6,137.6,136.4,128.9(2C),128.8(2C),128.5(2C),128.4(2C),128.2,128.1,79.7,
77.0,66.3,65.8,60.7,51.2,48.2,48.0,42.2,41.3,41.1,40.6,36.8,31.8,30.8,30.7,29.5,29.4,26.5,24.5,23.5,22.6,27.0,
20.2,19.7,18.7,15.6,17.9,14.6,14.4,14.1;MS (ESI):916.7(M+Na+);HRMS (MALDI/DHB) calcd.for (C49H75N5O10+Na+):916.5412,found:916.5426.
To a suspension of Pd/C (10%,10 mg) in CH3OH was added a solution of 18(100 mg,0.123 mmol) in CH3OH (3 mL).After the resulting mixture was stirred at room temperature under H2 atmosphere (1 atm) for 3 h,the catalyst was filtered off and the filtrate was concentrated in vacuo to give the crude deprotected product (74 mg,yield 99%),which was used in next step without further purification.The crude deprotected product was suspended in anhydrous CH3CN (120 mL,1.0 × 10-3 mol/L) and cooled to 0 ℃,pentafluorophenyldiphenylphosphinate (FDPP) reagent (85 mg,0.221 mmol) was added in one portion and stirred for 20 min at the same temperature.Then DIPEA (76 μL,0.435 mmol) was slowly added over a period of 30 min.After the addition was over,the mixture was allowed to warm to room temperature and stirred for 26 h.The mixture was concentrated in vacuo,and the residue was purified by RP C18 HPLC with 70% aqueous CH3CN (Sepax-tech Amethyst C18 semipreparative column,10 mm × 150 mm,2 mL/min,refractive index detection) to give emericellamide B (39 mg,yield 55% over two steps).[α]D25 -43.6(c 0.11,MeOH){lit.[α]D25 -34(c 0.076,MeOH)[10];[α]D25 -31(c 0.066,MeOH)[15];[α]D30-34.1(c 0.12,MeOH)[18]};IR (film,cm-1):υmax 3309,2931,1756,1632,1554;1H NMR (600 MHz,DMSO-d6)δ 8.27-7.98(m,2H),7.87(d,1H,J=7.51 Hz),7.49(bs,1H),7.34(d,1H,J=6.72 Hz),4.90(dd,1H,J=2.40,9.90 Hz),4.30(dd,1H,J=5.58,17.40 Hz),4.10-3.94(m,4H),3.63(dd,1H,J=5.58,17.40 Hz),2.87(dq,1H,J=7.13,10.24 Hz),1.89-1.84(m,1H),1.83-1.79(m,1H),1.62-1.50(m,4H),1.24(d,J=7.32 Hz,3H),1.21(d,3H,J=6.72 Hz),1.24-1.08(m,10H),1.06-0.98(m,1H),0.92-0.75(m,25H);13C NMR (150 MHz,DMSO-d6)δ 173.4,171.7,171.6,171.5,171.2,169.2,76.3,60.5,55.4,52.2,48.6,47.7,42.9,41.5,41.2,37.3,31.8,30.7,30.6,29.5,29.2,
26.6,25.0,23.6,22.6,21.3,19.9,19.5,19.2,18.8,16.8,14.8,14.5,14.1;MS (ESI):674.5(M+Na+);HRMS (MALDI/DHB) calcd.for (C34H61N5O7+Na+):674.4469,found:674.4474.
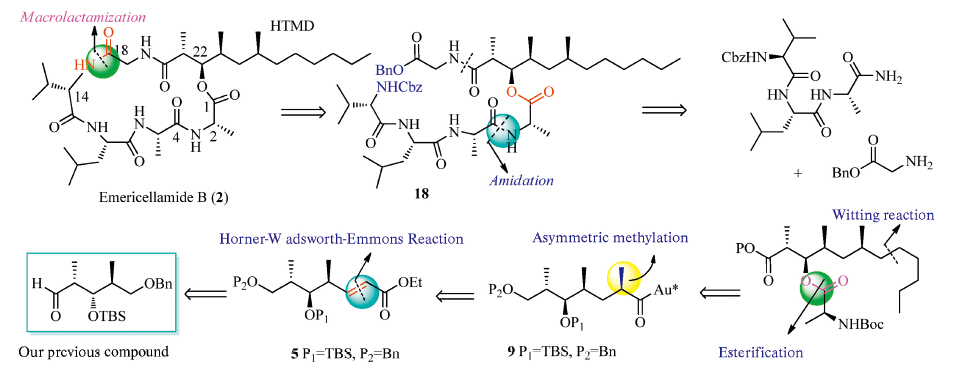
The retrosynthetic analysis of emericellamide B is shown in Scheme 1.Due to a highlymethylated (2R,3R,4S,6S)-3-hydroxy-2,4,6-trimethyldodecanoic acid (HTMD) unit in its structure the most critical step is the connection of the sterically hindered units (HTMD),with the other peptide-amino acid moiety.Yamaguchi protocol[23, 24]and condensation reaction between C-22 and C-1 were found to be ineffective[14, 15, 16, 17, 18].The goal of initial synthetic efforts is to achieve a general method for preparation of HTMD using cheapest material,and the esterification of small amino-acid with HTMD is the key task.In addition,convenient removal of the protecting groups in all these units would also have to be taken into account.

|
Download:
|
| Scheme. 1.Strategic bond disconnections of emericellamide B (2). | |
{kind=link}
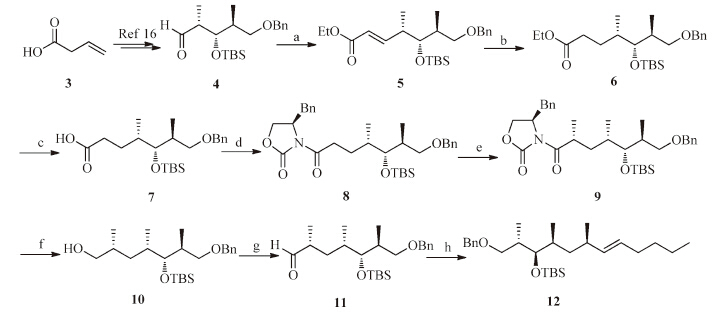
Scheme 2 shows the synthesis of the HTMD moiety of emericellamide B.The aldehyde 4 could be conveniently achieved from vinylacetic acid 3 using our previous method[16],and the (2R,3R,4S,6S)-3-hydroxy-2,4,6-trimethyldodecanoic acid (HTMD) was prepared using the following sequence.The Horner-Wadsworth-Emmons (HWE) reaction[25, 26]of aldehyde 4 with triethylphosphonoacetate in the presence of sodiumhydride predominantly afforded olefin 5 in E-form (The ratio of Z/E was 2:98) in 98% combined yield,and the minor Z-isomer could be easily separated by chromatography on silica gel.Reduction of 5 with sodium borohydride in the presence of nickel chloride[27]gave the ester 6 in 97% yield{[α]D25-14.7(c 0.82,CHCl3)}.In order to stereoselectively introduce methyl group into the α-position of carboxylic acid 7,Evans'methodwas chosen in our case.After the ester 6 was hydrolyzed to carboxylic acid 7 in excellent yield in the presence of LiOH.The reaction with (R)-4-benzyloxazolidin-2-one through the activated anhydride with pivaloyl chloride at -78 ℃ led to the amide 8{[α]D25 -24.2(c 5.34,CHCl3)}in 87% yield.The subsequent asymmetric methylation afforded 9 as the only desirable diastereomer in 74% yield[28, 29]{[α]D25 -50.7(c 1.15,CHCl3)}.The optimal conditions entailed the slow addition of 3 equiv.of methyl iodide over 30 min,and the reaction temperature was kept at -78 ℃ for 3 h to avoid the formation of a-dimethylated side-product.Removal of the Evan's auxiliary[30](LiBH4,MeOH) and Swern oxidation[31, 32](DMSO,(COCl)2) produced aldehyde 11,which was subjected to treatment with pentyltriphenylphosphonium bromide in the presence of n-BuLi to afford benzylic ether 12{[α]D25 -5.07(c 4.48,CHCl3)}in three steps with 83% yield.

|
Download:
|
| Scheme. 2.Synthesis of benzylic ether 12.Reagents and conditions:(a) triethylphosphonoacetate,NaH,THF,0 ℃ 1 h,98%;(b) NiCl2·6H2O,NaBH4,MeOH,THF,0 ℃,1 h,97%;(c) LiOH·H2O,THF/H2O,60 ℃,98%;(d)(R)-4-benzyloxazolidin-2-one,PivCl,Et3N,LiCl,THF,-78 ℃ to r.t.,overnight,87%;(e) NaHMDS,MeI,THF,-78 ℃ to r.t.,overnight,74%;(f) LiBH4,MeOH,THF,93%;(g) DMSO,(COCl)2,-78 ℃,30 min,then 10,-78 ℃ to -40 ℃,3 h;Et3N,-78 ℃ to r.t.,1 h,92%;(h) pentyltriphenylphosphonium bromide,n-BuLi,THF,r.t.,3 h,97%. | |
{kind=link}
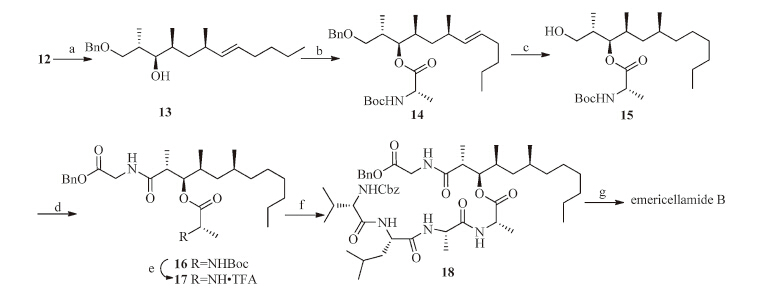
It was found that the Yamaguchi method for the macrocylization of emericellamide A was inefficient[14],most likely due to the steric hindrance in ex-macrocylization product.Furthermore,we have demonstrated that the macrolactonization of C-2 and C-4 was also inefficient in total synthesis of emericellamide A[16].Therefore,the esterification of less hindered secondary hydroxyl in 13 with N-Boc-Ala-OH was considered to form first in our synthetic strategy (Scheme 3).The coupling reaction of alcohol 13 with N-Boc-Ala-OH in the presence of EDC and DMAP in CH2Cl2 at 0 ℃ gave ester 14 in 60% yield,with partial epimerization at C-2 of product 14(13:1 ratio)[33],while the starting material could be recovered.It was worth mentioning that the minor epimer of 14 could be easily removed by chromatography on silica gel to give enatiopure 14{[α]D25 -11.3(c 0.56,CHCl3)}.Hydrogenation (Pd/C) of the ester 14 gave the alcohol 15 in quantitative yield.The crude acid,which was obtained through oxidation (RuCl3-NaIO4) of 15[34]was condensed with H-Gly-OBn in the presence of EDC/HOBt to produce 16 in 88% yield (three steps).Deprotection of N-Boc group (TFA,CH2Cl2) of 16 gave salt 17,which was coupled with N-Cbz-Val-Leu-Ala-OH under the same conditions as above to give polypeptide 18 in 65% yield (two steps).Removal of both Bn and Cbz-protecting groups (Pd/C,MeOH) led to free acid and amine in one pot,which was subjected to macrolactamization by treatmentwith pentafluorophenyl diphenylphoophinate (FDPP) and DIPEA under highly diluted solution (10-3 mol/L) of the substrate in CH3CN at room temperature to give emericellamide B in 55% yield (two steps).The crude emericellamide B was further purified by RP C18 HPLC with 70% aqueous CH3CN (Sepax-tech Amethyst C18 semipreparative column,10 mm × 150 mm,2 mL/min,refractive index detection){[α]D25 -43.6(c 0.11,MeOH);lit.[α]D25 -34(c 0.076,MeOH)[10];[α]D25 -31(c 0.066,MeOH)[15];[a]D 30 -34.1(c 0.12,MeOH)[18]}.In spite of the slightly different optical rotation of our synthetic emericellamide B,the spectroscopic data for our synthetic sample agreed well with that of the natural product[10].

|
Download:
|
| Scheme. 3.Synthesis of emericellamide B (2).Reagents and conditions:(a) MeOH/HCl,0 ℃ to r.t.,3 h,quantitative;(b) N-Boc-Ala-OH,EDCI,DMAP,CH2Cl2,0 ℃ to r.t.,6 h,60%,90%(starting material was recovered);(c) Pd/C (10%),H2,MeOH,r.t.,4 h;(d)(i) RuCl3·xH2O (cat.),NaIO4;(ii) HOBt,EDCI,-15 ℃,1.5 h;then H-Gly-OBn,DMF,-15 ℃ to r.t.,overnight,88%(three steps);(e) TFA,CH2Cl2,r.t.,2 h;(f) HOBt,N-Cbz-Val-Ile-Ala-OH,EDCI,-15 ℃,1.5 h;then 17,DMF,-15 ℃ to r.t.,overnight,65%(two steps);(g)(i) Pd/C (10%),H2,MeOH,r.t.,3 h;(ii) FDPP,DIPEA,CH3CN,0 ℃ to r.t.,26 h,55%(two steps). | |
{kind=link}
In conclusion,we have achieved a convenient method for total synthesis of emericellamide B.Our strategy demonstrated the preparation of the nonribosomal moieties of emericellamide B starting from cheap vinylacetic acid 3.Therefore,a general and efficient method for total synthesis of emericellamides and their analogues was established.Additionally,the concise synthesis could be beneficial for studying their structure-activity-relationship and their effects on human health,as well as providing enough material for exploring their possible antimicrobial mechanism and pharmacology. Acknowledgments
We thank the National Natural Science Foundation of China (Nos.21472022,21272041,21072034),Key Laboratory for Chemical Biology of Fujian Province for financial support.
Appendix A.Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.07.023.
| [1] | J.W. Blunt, B.R. Copp, R.A. Keyzers, M.H.G. Munro, M.R. Prinsep, Marine natural products, Nat. Prod. Rep. 31(2014) 160-258. |
| [2] | P. Zhang, X.M. Li, J.N. Wang, X. Li, B.G. Wang, Prenylated indole alkaloids from the marine-derived fungus Paecilomyces variotii, Chin. Chem. Lett. 26(2015) 313-316. |
| [3] | K.X. Zhan, W.H. Jiao, F. Yang, et al., Reniochalistatins A-E, cyclic peptides from the marine sponge Reniochalina stalagmitis, J. Nat. Prod. 77(2014) 2678-2684. |
| [4] | J. Kitagaki, G. Shi, S. Miyauchi, S. Murakami, Y. Yang, Cyclic depsipeptides as potential cancer therapeutics, Anti-Cancer Drugs 26(2015) 259-271. |
| [5] | J. Swathi, K. Narendra, K.M. Sowjanya, A.K. Satya, Marine fungal metabolites as a rich source of bioactive compounds, Afr. J. Biochem. Res. 7(2013) 184-196. |
| [6] | L.A. Salvador-Reyes, H. Luesch, Biological targets and mechanisms of action of natural products from marine cyanobacteria, Nat. Prod. Rep. 32(2015) 478-503. |
| [7] | W. Li, A. Schlecker, D. Ma, Total synthesis of antimicrobial and antitumor cyclic depsipeptides, Chem. Commun. (Cambridge, U.K.) 46(2010) 5403-5420. |
| [8] | D.B.M. Virupakshaiah, Docking of secondary metabolites derived frommarine fungi with Hsp90a protein in cancer treatment, J. Adv. Bioinf. Appl. Res. 5(2014) 92-96. |
| [9] | G.S. Bagavananthem Andavan, R. Lemmens-Gruber, Cyclodepsipeptides from marine sponges:natural agents for drug research, Mar. Drugs 8(2010) 810-834. |
| [10] | D.C. Oh, C.A. Kauffman, P.R. Jensen, W. Fenical, Induced production of emericellamides A and B from the marine-derived Fungus Emericella sp. in competing coculture, J. Nat. Prod. 70(2007) 515-520. |
| [11] | P. Marfey, Determination of D-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene, Carlsberg Res. Commun. 49(1984) 591-596. |
| [12] | J.M. Seco, E. Quinoa, R. Riguera, A practical guide for the assignment of the absolute configuration of alcohols, amines and carboxylic acids by NMR, Tetrahedron:Asymmetry 12(2001) 2915-2925. |
| [13] | Y.M. Chiang, E. Szewczyk, T. Nayak, et al., Molecular genetic mining of the Aspergillus secondary metabolome:discovery of the emericellamide biosynthetic pathway, Chem. Biol. (Cambridge, MA, U.S.) 15(2008) 527-532. |
| [14] | S. Ghosh, T.K. Pradhan, The first total synthesis of emericellamide A, Tetrahedron Lett. 49(2008) 3697-3700. |
| [15] | S. Li, S. Liang, W. Tan, Z. Xu, T. Ye, Total synthesis of emericellamides A and B, Tetrahedron 65(2009) 2695-2702. |
| [16] | J.Y. Ma, L.F. Xu, W.F. Huang, B.G. Wei, G.Q. Lin, Total synthesis of emericellamide A:a secondary metabolite of marine cyclic depsipeptide with antimicrobial properties, Synlett (2009) 1307-1310. |
| [17] | D.K. Mohapatra, S. Samad Hossain, S. Dhara, J.S. Yadav, Application of desymmetrization protocol for the formal total synthesis of emericellamide B, Tetrahedron Lett. 51(2010) 3079-3082. |
| [18] | T.K. Pradhan, K.M. Reddy, S. Ghosh, Total synthesis of emericellamides A and B, Tetrahedron:Asymmetry 24(2013) 1042-1051. |
| [19] | C.M. Si, W. Huang, Z.T. Du, B.G. Wei, G.Q. Lin, Diastereoconvergent synthesis of trans-5-hydroxy-6-substituted-2-piperidinones by addition-cyclization-deprotection process, Org. Lett. 16(2014) 4328-4331. |
| [20] | C.M. Si, Z.Y. Mao, H.Q. Dong, et al., Divergent method to trans-5-hydroxy-6-alkynyl/alkenyl-2-piperidinones:syntheses of (-)-epiquinamide and (+)-swainsonine, J. Org. Chem. 80(2015) 5824-5833. |
| [21] | J.Y. Ma, W. Huang, B.G. Wei, Asymmetric synthesis of (E)-dehydroapratoxin A, Tetrahedron Lett. 52(2011) 4598-4601. |
| [22] | W. Huang, R.G. Ren, H.Q. Dong, B.G. Wei, G.Q. Lin, Diverse synthesis of marine cyclic depsipeptide lagunamide A and its analogues, J. Org. Chem. 78(2013) 10747-10762. |
| [23] | J. Inanaga, K. Hirata, H. Saeki, T. Katsuki, M. Yamaguchi, A rapid esterification by mixed anhydride and its application to large-ring lactonization, Bull. Chem. Soc. Jpn. 52(1979) 1989-1993. |
| [24] | X.S. Chen, S.J. Da, L.H. Yang, et al., A new convenient asymmetric approach to herbarumin III, Chin. Chem. Lett. 18(2007) 255-257. |
| [25] | L. Horner, H. Hoffman, H.G. Wippel, G. Klahre, Phosphorus organic compounds. XX. Phosphine oxides as reagents for olefin formation, Chem. Ber. 92(1959) 2499-2505. |
| [26] | J. Gao, Y.H. Guo, Y.P. Wang, X.J. Wang, W.S. Xiang, A novel and efficient route for the preparation of atorvastatin, Chin. Chem. Lett. 22(2011) 1159-1162. |
| [27] | D. Dhawan, S.K. Grover, Facile reduction of chalcones to dihydrochalcones with sodium borohydride/nickel(II) system, Synth. Commun. 22(1992) 2405-2409. |
| [28] | D.A. Evans, M.D. Ennis, D.J. Mathre, Asymmetric alkylation reactions of chiral imide enolates. A practical approach to the enantioselective synthesis of a-substituted carboxylic acid derivatives, J. Am. Chem. Soc. 104(1982) 1737-1739. |
| [29] | J.C. Liu, Y.S. Yang, R.Y. Ji, A convenient method for the asymmetric synthesis of KAD-1229, Chin. Chem. Lett. 16(2005) 430-432. |
| [30] | T.D. Penning, S.W. Djuric, R.A. Haack, et al., Improved procedure for the reduction of N-acyloxazolidinones, Synth. Commun. 20(1990) 307-312. |
| [31] | A.J. Mancuso, D. Swern, Activated dimethyl sulfoxide:useful reagents for synthesis, Synthesis (1981) 165-185. |
| [32] | Y. Wang, Z.Z. Liu, Y.F. Tang, S.Z. Chen, An efficient synthetic strategy for construction of functionalized pentacyclic skeleton of ecteinascidin-saframycin alkaloids, Chin. Chem. Lett. 17(2006) 853-856. |
| [33] | L.H. Shen, H.Y. Li, H.X. Shang, et al., Synthesis and cytotoxic evaluation of new colchicine derivatives bearing 1,3,4-thiadiazole moieties, Chin. Chem. Lett. 24(2013) 299-302. |
| [34] | P.H.J. Carlsen, T. Katsuki, V.S. Martin, K.B. Sharpless, A greatly improved procedure for ruthenium tetroxide catalyzed oxidations of organic compounds, J. Org. Chem. 46(1981) 3936-3938. |