 2015, Vol.26
2015, Vol.26 


 , Yang Hea, Yu Wanga, Jing-Tao Lüc,d, Hao Tangf, Lian-Mao Penga, Shi-Min Houa, Kai Wue , Yong-Feng Wanga,d
, Yang Hea, Yu Wanga, Jing-Tao Lüc,d, Hao Tangf, Lian-Mao Penga, Shi-Min Houa, Kai Wue , Yong-Feng Wanga,d
b Department of Theoretical Chemistry, Miria-Curie Skłodowska University, PL. M. C. Skłodowskiej 3, 20-031, Lublin, Poland;
c School of Physics, Huazhong University of Science and Technology, Wuhan 430074, China;
d Beida Information Research(BIR), Tianjin 300457, China;
e BNLMS, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China;
f Groupe Matériaux Crystallins sous Contrainte, CEMES-CNRS, Boîte Postale 94347, 31055 Toulouse, France
Self-similar fractal structures exist widely in nature,from the well-known snowflakes to the complicate Saturn’s rings [1]. Exploration of these fractals is not only fundamentally important in science and engineering,but also interesting in esthetics. It has been theoretically predicted that fractal frameworks comprising Sierpiński-triangle (ST) units could exhibit peculiar mechanical,electronic,and magnetic properties [2]. In this context,various fractals,including a first-generation ST,have been successfully synthesized in solutions through sophisticated design [3, 4, 5, 6, 7, 8]. However,synthesis of larger fractals usually encounters difficulties due to their poor solubility. Surface-assisted synthesis facilitates the formation of much larger and more complicated fractal structures. Molecular and atomic dendritic fractals with irregular shapes on surfaces could form through the diffusion-limited aggregation (DLA) process at conditions far from equilibrium [9, 10]. After annealing,they usually change to close-packed ordered structures.
Very recently,we reported the formation of extended and defect-free STs on the Ag(1 1 1) surface under ultrahigh vacuum conditions after cooling the substrate from liquid to solid nitrogen temperature (77-65 K) [11]. The STs stabilized by weak cyclic halogen bonds are only stable up to 65 K. A prerequisite for interesting applications of STs is their thermal stability,which can be improved by using stronger coordination or covalent bonds. The former,incorporating metals to fractals,is particularly attractive for applications in gas separation,catalysis and as sensing [12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23]. However,this is extremely challenging due to the fact that metals and ligands have a strong tendency to form low dimensional crystals or random networks on surfaces.
To construct metal-organic (MO) STs,120° V-shaped molecules and three-fold nodes are essential [11, 24]. Surface coordination chemistry demonstrated that the cyano (-CN) group and Fe could form three-fold bonding on single-crystal surfaces [25, 26, 27, 28]. Therefore,4,4"-dicyano-1,1':3',1"-terphenyl (C3PC,Fig. 1a) molecules and Fe atoms were used in this study to fabricate metalorganic Sierpiński triangles (MOSTs) on surfaces. As expected,a series of defect-free fractal MOST crystals with the C3v point group were successfully prepared on Au(1 1 1).

|
Download:
|
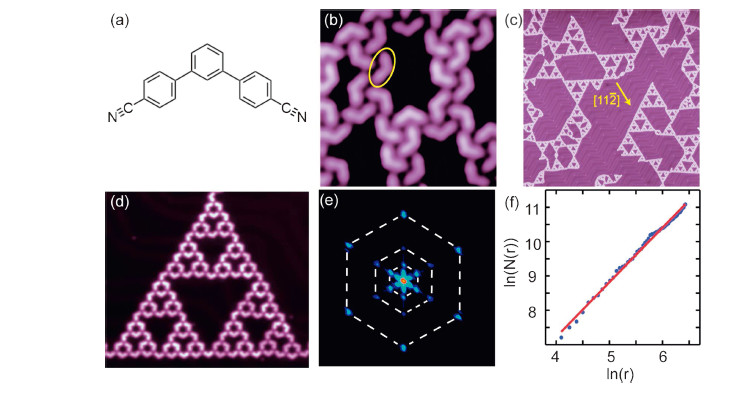
| Fig. 1.(a) Optimized structure of C3PC (H atoms not shown). (b) STM topography of the superstructure formed by C3PC molecules (7.2 nm ×7.2 nm; sample voltage U = 1 V, current I = 30 pA). (c) MOSTs formed by depositing Fe onto one molecule covered surface and annealing (180 nm ×180 nm; sample voltage U = 1 V, current I = 17 pA). (d) High-resolution STM image of a typical Sierpiński triangle Fe-ST-3 (29.3 nm ×25.6 nm; U = -0.05 V, CH). (e) Fast Fourier transformation of (c). (f) Plot of log(N(r)) versus ln(r) giving the fractal dimension d = 1.59. Crystal direction [1 1 2] was marked by the yellow arrow according to the herringbone reconstruction on Au(1 1 1) surface. | |
{kind=link}
The experiments were carried out with a Unisoku scanning tunneling microscope (STM) at a temperature of 4.3 K with a base pressure of 10-10 Torr. The single crystalline Au(1 1 1) surface was cleaned by repetitive cycles of Ar ion sputtering and annealing at 400°C. Polycrystalline Pt/Ir tips were annealed first in the vacuum chamber and treated by dipping into substrates gently. C3PC molecules were thermally deposited on the substrates held at room temperature from a Ta boat heated by direct current. And then Fe was evaporated from another Ta boat to the moleculecovered surfaces. After annealing at around 100°C for 10 min,the samples were transferred to STM and imaged at liquid helium temperature. The images were processed using software WSxM.
3. Results and discussionFig. 1b shows a constant-current (CC) scanning tunneling microscope (STM) image taken after deposition of the C3PC molecules on Au(1 1 1). The ellipse in the figure encloses a single molecule,which looks like a boomerang. Molecules form chainlike structures through the multiple weak hydrogen bonds between the cyano group and the H-atoms of the phenyl rings. After thermally sublimating Fe onto the C3PC covered Au(1 1 1) surface and annealing at around 100°C for 10 min,chain-like structures vanished and a series of equilateral triangles appeared on the substrate (Fig. 1c). The newly formed superstructures should be bonded through coordination interaction between Fe atoms and C3PC molecules. As expected,Fe-N bonds are three-fold symmetric. High-resolution STM image (Fig. 1d) obtained in constant height (CH) mode reveals detailed characteristics of one triangle. It is self-similar,depicting the properties of STs. Therefore,we denote them as Fe-ST-n,where n is order of the Sierpiński triangle and n = 0,1,2,3,4 in this article. Under the condition of lower coverage and slower annealing,MOSTs of higher orders are possible to be identified experimentally.
To demonstrate the symmetry of Fig. 1d,its fast Fourier transformation was performed. Several self-similar hexagonal shells are shown in Fig. 1e. The outer shell corresponds to the smallest period inside one triangle,distance between one molecule (node) and its nearest neighbor (node). Its radius a1 equals 1.69 nm,which is much larger than that of a free molecular (1.44 nm) owing to occupation of the metal atom between each pair of molecules. The second shell with a radius a2 = 3.59 nm,corresponds to the packing style of Fe-ST-1s inside the Fe-ST-3,while the third shell (a3 = 7.32 nm) corresponds to Fe-ST-2s. The ratio a2/a1 = a3/a2 = 0.5 in k-space coincides with that of the STs in real space.
Hausdorff dimension,defined as d = - limR→0logRN(R),is another important parameter describing a fractal. Here,N is the smallest number to cover the set of all points in object with sphere of radius R reaching to 0. But sometimes Hausdorff dimension is difficult to obtain,so the box-counting dimension is adopted instead. We applied the linear fit of ln(N(r)) versus r and acquired d = 1.59,almost the same as the ideal dimension of Sierpiński triangle d = log2 3 = 1.58. The slight deviation may arise from the fact that molecules in the observed Fe-ST-3 (Fig. 1d) possess subtle geometric features,like vertices and width. With increase of the STs order,the deviation further decreases.
The STs of an order up to 3 and an incomplete Fe-ST-4 were obtained in experiments and shown in Fig. 2a-e. The down-right ST-3 inside ST-4 was missing and patched with modeled molecules. For arbitrary ST-n,the molecular (An) and atomic (Mn) numbers are:


|
Download:
|
Fig. 2.(a–e) STM images of Fe-ST-n with n equals 0, 1, 2, 3, 4, respectively. (f–g) The formation of Fe-ST-n. One ST-(n - 1) is fixed, while the other two are translated by 2n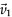 and 2n and 2n . Then three assisted molecules are added between them in the structure. The molecular, atomic numbers and area of the largest pore (colored in gray in f–g) of STs are marked below each Fe-ST-n. Scanning conditions: (a) 4.9 nm ×4.5 nm, U = 0.01 V, CH; (b) 6.3 nm ×6.3 nm, U = 0.01 V, CH; (c) 13.3 nm ×13.0 nm, U = 0.01 V, CH; (d) 27.9 nm ×25.4 nm, U = 0.1 V, CH; (e) U = 1 V, I = 17.7 pA. . Then three assisted molecules are added between them in the structure. The molecular, atomic numbers and area of the largest pore (colored in gray in f–g) of STs are marked below each Fe-ST-n. Scanning conditions: (a) 4.9 nm ×4.5 nm, U = 0.01 V, CH; (b) 6.3 nm ×6.3 nm, U = 0.01 V, CH; (c) 13.3 nm ×13.0 nm, U = 0.01 V, CH; (d) 27.9 nm ×25.4 nm, U = 0.1 V, CH; (e) U = 1 V, I = 17.7 pA.
|
|
{kind=link}
These two numbers were also presented in Fig. 2. It should be pointed out that the molecules attached to vertices of STs are not counted here. The areas of the largest pores of Fe-STs (colored in gray) are marked in the figure,which range from 0.7 to 80.4 nm2. To simplify the structure of STs,we replace the C3PC molecule with a 120° polyline according to geometric configuration. A blue circle represents a metal atom,and its coordination number on the surface is 3. Each ST-n can be formed by three STs-(n - 1) and three assisted molecules (purple). There into,one ST-(n - 1) is fixed,while the other two are translated by 2n and 2n,respectively. and ,defined in Fig. 2f,are identical except for the vector direction. Then,three assisted molecules are added to the structure to saturate the coordination.
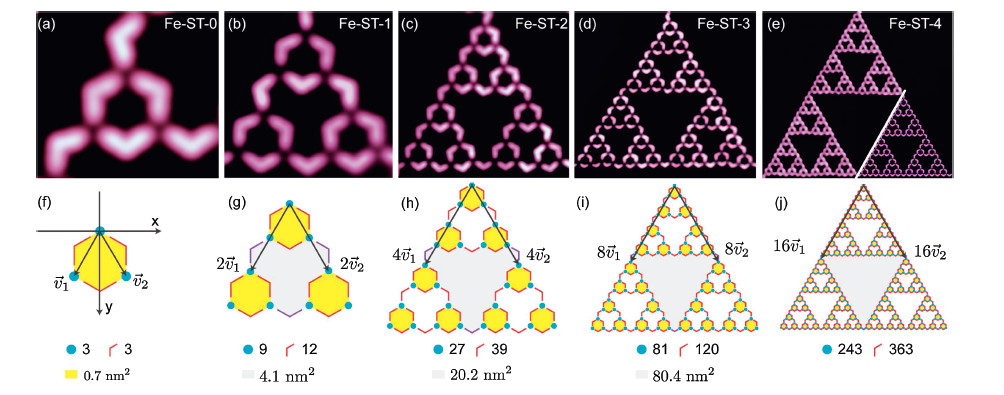
The Fe atom is elusive in most STM images. To elaborate the rationale,density functional theory (DFT) calculations were performed. Fig. 3a shows the optimized structure of one Fe atom and three C3PC molecules adsorbed on Au(1 1 1). The Fe atom locates on a fcc hollow site,and the Fe-N bond length is about 1.88Å ,which is consistent with other DFT calculations. The terphenyl adopted a twisted conformation with a dihedral angle of about 20°,and an average distance of 3.50Å to the Au(1 1 1) surface. The Fe atom is 1.62Å farther away from the tip compared to the C3PC molecules (Fig. 3b),which might result in weaker coupling to the tip and invisibility in the STM image. Occasionally,the STM tip was modified by residual gases in vacuum,and then its resolution was greatly enhanced. In such a situation,the Fe atom could be clearly observed in the image,as shown in Fig. 3c.

|
Download:
|
| Fig. 3.(a, top view) and (b, side view) are optimized structures comprising one Fe atom and three C3PC molecules on Au(1 1 1). (c) High-resolution STM image of a Fe-ST-2 obtained by a molecule modified tip (11.7 nm ×15.7 nm; 5 mV, CH). | |
{kind=link}
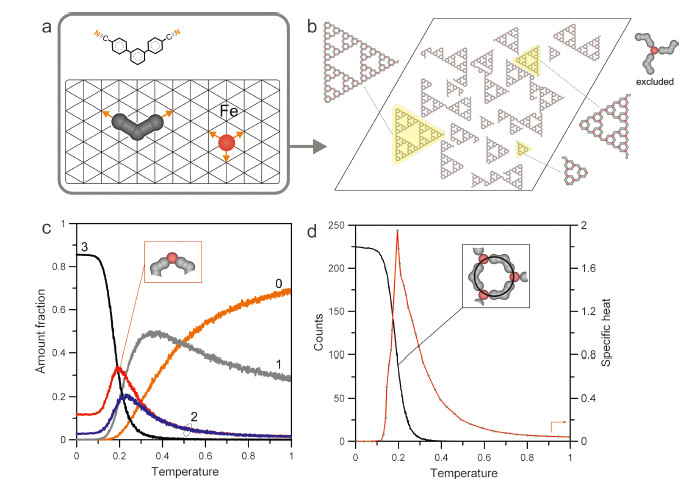
To understand the formation process of MOSTs,we carried out Monte Carlo [24, 29, 30] simulations performed for a mixture of 1050 linker molecules and 700 metal atoms whose mapping on a triangular lattice is shown in Fig. 4a. A detailed description of the simulation algorithm and basic assumptions of the model can be found in the Supporting information. As the MOSTs observed in the experiment exhibit an unusual ability to self-correction through elimination of the windmill nodes (Fig. 4b),we focus on the theoretical results based on the assumption that these nodes are not possible to form. A possible origin of the absence of windmill nodes in the adsorbed phase is given in Fig. S1 in Supporting information.

|
Download:
|
| Fig. 4.(a) Mapping of the components of the metal-organic self-assembly onto a triangular lattice representing the Au(1 1 1) surface. The orange arrows indicate the interaction directions assumed for each species. For the iron atom an interaction pattern leading to the formation of a three-fold coordination node is presented. (b) Snapshot of the metal-organic overlayer comprising 1050 linker molecules and 700 Fe atoms (3:2 ratio) adsorbed on a 200 by 200 triangular lattice. In the simulation, creation of threefold windmill nodes (of which one surface enantiomer is shown in the right upper corner) was prohibited. (c) Temperature dependence of the relative abundance of metal coordination centers with a given number (0–3) of attached linker molecules. (d) Number of hexagonal pores occurring in the aggregates from panel (a) and the corresponding specific heat plotted as functions of temperature. | |
{kind=link}
Fig. 4b shows a representative snapshot of the adsorbed overlayer in which STs of first,second and third generation can be clearly seen. The internal structure of these aggregates agrees perfectly with the corresponding experimental counterpart from Fig. 2. This proves that the simple MC model based on directional metal-linker interactions does lead to complex hierarchical structures which can indeed exist in reality. To gain deeper insight into the formation mechanism of the STs in Fig. 4c,we plotted the relative populations of metal centers with 0-3 attached linker molecules at different temperatures. The calculated dependencies are qualitatively similar to those obtained previously for the model,in which the formation of the windmill nodes was allowable [1]. In particular,we can observe that the formation of the STs at low temperatures is dictated by the rapid increase in the number of three-fold coordination nodes occurring at temperature equal to about 0.2 (see Supporting information for the definition of temperature). This transformation involves disappearance of the two-fold coordination nodes,some of which,however,persist at the target temperature ( < 0.1). Most of these nodes (see the red curve) are the corners of the STs (shown in the inset),but they can also be found at higher temperatures (>0.3) where the formation of triangular aggregates is highly ineffective.
A possible explanation of this effect would be the formation of cyclic oligomers comprising three linker molecules and three metal atoms,in which each metal atom is in twofold “corner” coordination (Fig. S2 in Supporting information). To test this hypothesis,we reckoned independently the number of the hexagonal units (pores) built of three linker molecules,as shown in the inset of Fig. 4d. The corresponding temperature dependence plotted in this figure indicates that the cyclic units are practically absent for temperatures higher than 0.3. However,as it follows from Fig. 4c,the twofold nodes which can potentially create these units (red) are quite highly populated at 0.3 (15%),which clearly shows a different and more diversified mechanism of the selfassembly of the STs at the initial stage. Here instead of a collective “cyclization reaction” and subsequent linkage of these basic hexagonal units,we are dealing with the formation of incomplete (open) pores (Fig. S3 in Supporting information) whose twofold coordinated metal atoms become threefold coordinated via the attachment of a next linker molecule. Formation of such nodes competes with pore closing,as it provides a more stable configuration for the nodal metal atom (three bonds). In consequence,the initial growth of the triangles occurs also due to the creation of nodes where the metal atoms reach maximum coordination (three) without undergoing through self-assembly of the cyclic oligomers. The dominating role of the three-fold nodes in the structure formation becomes evident when the corresponding specific heat curve from Fig. 4d is considered. The sharp peak which is an indication of the structural change from 2D gas phase to supramolecular phase reaches a maximum whose position on the temperature axis (~0.2) coincides with the onset of the fastest growth of the three-fold nodes (Fig. 4c) and pores (Fig. 4d).
4. ConclusionsMetal-organic Sierpiński triangles were successfully constructed by using 120° V-shaped 4,4"-dicyano-1,1':3',1"-terphenyl molecules and Fe atomsonAu(1 1 1). Full STswithan order up to 3 and an incomplete Fe-ST-4were experimentally obtained. Thewhole series of defect-free fractal crystals belong to the C3v point group. The structures were characterized in detail by low-temperature STM. DFT calculations were performed to understand the geometric and electronic structures of STs,revealing that the Fe atom is 1.62Å farther away from the tip compared to the C3PC molecules,and making the Fe atoms elusive in STMimages achieved by a metal tip. Monte Carlo simulations were carried out to unravel the formation process of surface-supported fractal crystals. It turns out that the three-fold nodes dominate the structure formation of fractal crystals.
AcknowledgmentsThis work was jointly supported by National Natural Science Foundation of China (Nos. 21373020,21403008,61321001,21433011,21522301,21133001,21333001,913000002),Ministry of Science and Technology (Nos. 2014CB239302,2013CB933404,2011CB808702),and the Research Fund for the Doctoral Program of Higher Education (No. 20130001110029).
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.08.006.
| [1] | B.B. Mandelbrot, The Fractal Geometry of the Nature, W.H. Freeman and Company, 1982. |
| [2] | E. van Veen, A. Tomadin, M.I. Katsnelson, S. Yuan, M. Polini, Transport and optical properties of an electron gas in a Sierpinski carpet,http://es.arxiv.org/abs/1504.00628. |
| [3] | G.R. Newkome, C. Shreiner, Dendrimers derived from 1 to 3 branching motifs, Chem. Rev. 110(2010) 6339-6442. |
| [4] | K.I. Sugiura, H. Tanaka, T. Matsumoto, T. Kawai, Y. Sakata, A Mandala-patterned bandanna-shaped porphyrin oligomer, C1224H1350N84Ni20O88, having a unique size and geometry, Chem. Lett. 28(1999) 1193. |
| [5] | G.R. Newkome, P. Wang, C.N. Moorefield, et al., Nanoassembly of a fractal polymer:a molecular "Sierpiński hexagonal gasket", Science 312(2006) 1782-1785. |
| [6] | K. Fujibayashi, R. Hariadi, S.H. Park, E. Winfree, S. Murata, Toward reliable algorithmic self-assembly of DNA tiles:a fixed-width cellular automaton pattern, Nano Lett. 8(2008) 1791-1797. |
| [7] | R. Sarkar, K. Guo, C.N. Moorefield, et al., One-step multicomponent self-assembly of a first generation Sierpiński triangle:from fractal design to chemical reality, Angew. Chem. Int. Ed. 53(2014) 12182-12185. |
| [8] | M. Wang, C. Wang, X.Q. Hao, et al., Hexagon wreaths:self-assembly of discrete supramolecular fractal architectures using multitopic terpyridine ligands, J. Am. Chem. Soc. 136(2014) 6664-6671. |
| [9] | H. Röder, E. Hahn, H. Brune, J.P. Bucher, K. Kern, Building one- and two-dimensional nanostructures by diffusion-controlled aggregation at surfaces, Nature 366(1993) 141-143. |
| [10] | H. Brune, C. Bomainczyk, H. Röder, K. Kern, Mechanism of the transition from fractal to dendritic growth of surface aggregates, Nature 369(1994) 469-471. |
| [11] | J. Shang, Y. Wang, M. Chen, et al., Assembling molecular Sierpiński triangle fractals, Nat. Chem. 7(2015) 389-393. |
| [12] | S.S.Y. Chui, S.M.F. Lo, J.P.H. Charmant, A.G. Orpen, I.D. Williams, A chemically functionalizable nanoporous material[Cu3(TMA)2(H2O)3] n, Science 283(1999) 1148-1150. |
| [13] | H. Li, M. Eddaoudi, M. O'Keeffe, O.M. Yaghi, Design and synthesis of an exceptionally stable and highly porous metal-organic framework, Nature 402(1999) 276-279. |
| [14] | C.E. Wilmer, M. Leaf, C.Y. Lee, et al., Large-scale screening of hypothetical metalorganic frameworks, Nat. Chem. 4(2012) 83-89. |
| [15] | E.D. Bloch, W.L. Queen, R. Krishna, et al., Hydrocarbon separations in a metalorganic framework with open iron(II) coordination sites, Science 355(2012) 1606-1610. |
| [16] | Z.R. Herm, B.M. Wiers, J.A. Mason, et al., Separation of hexane isomers in a metalorganic framework with triangular channels, Science 340(2013) 960-964. |
| [17] | Y. Inokuma, S. Yoshioka, J. Ariyoshi, et al., X-ray analysis on the nanogram to the microgram scale using porous complexes, Nature 495(2013) 461-466. |
| [18] | H. Furukawa, K.E. Cordova, M. O'Keeffe, O.M. Yaghi, The chemistry and applications of metal-organic frameworks, Science 341(2013), http://dx.doi.org/10.1126/science.1230444. |
| [19] | P. Deria, J.E. Mondloch, O. Karagiaridi, et al., Beyond post-synthesis modification:evolution of metal-organic frameworks via building block replacement, Chem. Soc. Rev. 43(2014) 5896-5912. |
| [20] | X.W. Wang, H. Guo, M.J. Liu, X.Y. Wang, D.S. Deng, 2D naphthalenedisulfonate-cadmium coordination polymer with 2,4,5-tri(4-pyridyl)-imidazole as a co-ligand:structure and catalytic property, Chin. Chem. Lett. 25(2014) 243-246. |
| [21] | Y.X. Sun, W.Y. Sun, Influence of temperature on metal-organic frameworks, Chin. Chem. Lett. 25(2014) 823-828. |
| [22] | T.M. McDonald, J.A. Mason, X. Kong, et al., Cooperative insertion of CO2 in diamine-appended metal-organic frameworks, Nature 519(2015) 303-308. |
| [23] | X.X. Liu, Y. Wang, W.G. Tian, W. Yang, Z.M. Sun, Heterometallic zinc uranium oxyfluorides incorporating imidazole ligands, Chin. Chem. Lett. 26(2015) 641-645. |
| [24] | D. Nieckarz, P. Szabelski, Simulation of the self-assembly of simple molecular bricks into Sierpiński triangles, Chem. Comm. 50(2014) 6843-6845. |
| [25] | S. Steppanow, N. Lin, D. Payer, et al., Surface assisted assembly of 2D metalorganic networks that exhibit unusual threefold coordination symmetry, Angew. Chem. Int. Ed. 46(2007) 710-713. |
| [26] | U. Schlickum, R. Decker, F. Klappenberger, et al., Metal-organic honeycomb nanomeshes with tunable cavity size, Nano Lett. 7(2007) 3813-3817. |
| [27] | U. Schlickum, F. Klappenberger, R. Decker, et al., Surface-confined metal-organic nanostructures from Co-directed assembly of linear terphenyl-dicarbonitrile linkers on Ag(111), J. Phys. Chem. C 114(2010) 15602-15606. |
| [28] | J. Xu, Q.D. Zeng, Construction of two-dimensional (2D) H-bonded supramolecular nanostructures studied by STM, Chin. Chem. Lett. 24(2013) 177-182. |
| [29] | D. Nieckarz, P. Szabelski, Understanding pattern formation in 2D metal-organic coordination systems on solid surfaces, J. Phys. Chem. C 117(2013) 11229-11241. |
| [30] | D. Frenkel, B. Smit, Understanding Molecular Simulation from Algorithms to Applications, Academic Press, 2002. |