 2015, Vol.26
2015, Vol.26 



b Department of Pharmacy, General Hospital of PLA Chengdu Military Area Command, Chengdu 610083, China
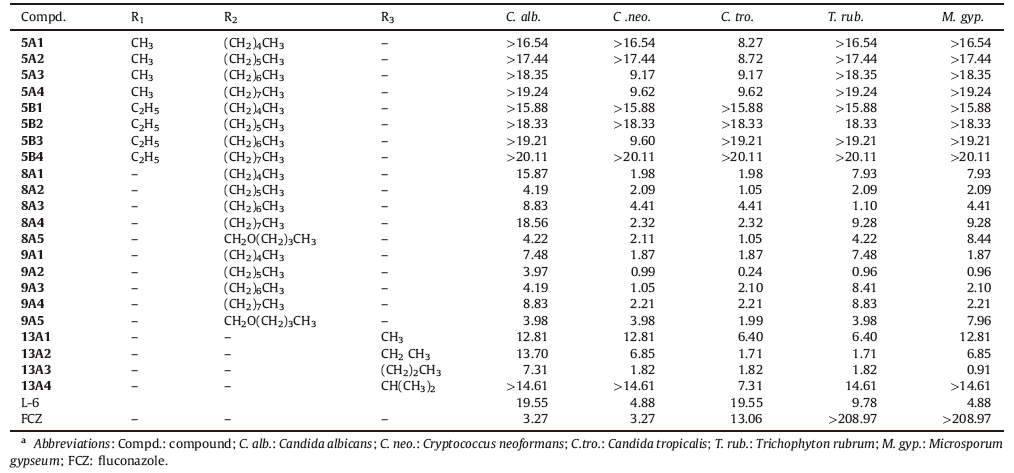
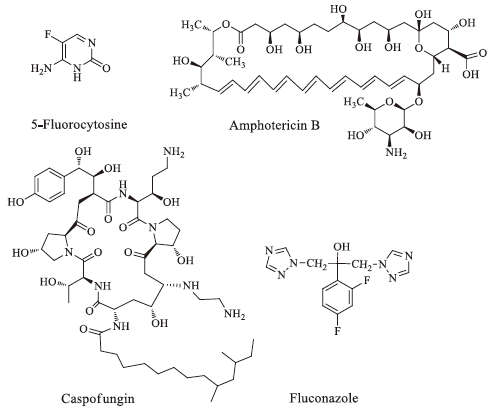
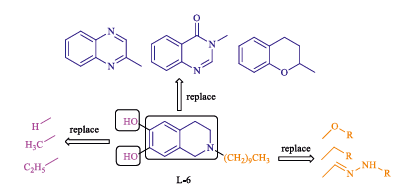
During the past several years, the incidence of life-threatening invasive and systemic fungal infections has increased dramatically due to an increase in the number of immunocompromised hosts, such as patients undergoing anticancer chemotherapy or organ transplants and patients with AIDS [1, 2]. Several antifungal drugs such as azoles, amphotericin B [3], 5-fluorocytosine [4], and caspofungin [5] have been developed to reduce the impact of fungal diseases (Fig. 1). Azoles, especially triazole agents, are used widely and effectively as first-line agents for the treatment of fungal infections in the clinic. However, the increasing administration of antifungal agents has led to fungal resistance [6, 7], and the currently available antifungal drugs do not meet the requirements of clinical treatments for complex infections. Therefore, new antifungal drugs are a major focus of drug development. Lanosterol 14a-demethylase (CYP51) is a key enzyme in fungal sterol biosynthesis [8]. Azole antifungals are competitive inhibitors of CYP51 that are used clinically [9]; however, cases of fatal hepatotoxicity with azole drugs have been reported due to the coordination of azoles with the heme groups of cytochrome P450 enzymes [10]. Therefore, the search for new non-azole CYP51 inhibitors is important, and this area has been a focus of our research in recent years. Three-dimensional models of CYP51 have been constructed using the homology modeling technique [11- 13]. Among our reported non-azole lead compounds, the molecules based on the isoquinoline scaffold showed potent antifungal activity, and L-6 was the most potent compound that interacted with the residues of fungal CYP51 apoprotein without binding to the heme (Figs. 1 and 2) [14, 15]. A series of compounds were designed and synthesized to investigate the structure- activity relationships of non-azole antifungal compounds. First, we replaced the isoquinoline scaffold with quinazolinone, benzopyran or quinoxaline in order to investigate the antifungal activity of the core scaffold structure. A long alkyl side chain was introduced at the position 3 of the quinazolinone, the position 2 of the quinoxaline or the benzopyran ring, in order to interact with the hydrophobic S3 pocket of CYP51. As a result, a series of 3- substituted quinazolinone compounds, 2-substituted quinoxaline compounds and 2-substituted benzopyran compounds (Fig. 2) were designed and synthesized, and their antifungal activity was evaluated in vitro.
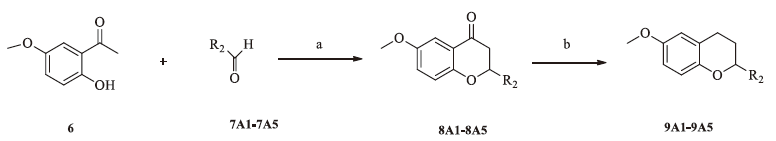
2. ExperimentalThe chemical synthesis of heterocyclic compounds is outlined in Schemes 1-3. First, treatment of substituted 2-aminobenzoic acid 1A-1B with propionic acid anhydride 2 afforded compounds 3A-3B [16, 17, 18]. Then, intermediates 3A-3B were reacted with different alkylamines in dry dimethylformamide (DMF) to afford the targeted compounds 5A-5B. Compounds 8A1-8A5 were synthesized according to a reported procedure [19, 20] by heating ethanolic mixtures to 160-170 °C or using microwave (MW) irradiation for 1 h in the presence of diisopropylamine (DIPA) as a base. Reduction of 8A1-A5 with lithium aluminum hydride (LiAlH4) in an ice bath yielded the targeted compounds 9A1-A5.

|
Download:
|
| Fig. 1.Antifungal drugs. | |

|
Download:
|
| Fig. 2.The targeted compounds. | |
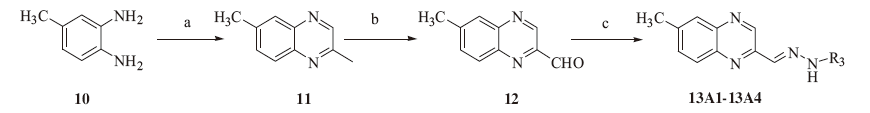
In order to generate useful intermediates, condensation of 1, 2- diaminobenzene 10 with pyruvaldehyde gave 2-methylquinoxaline 11 in good yields [21]. A good method for preparing the quinoxaline-2-carboxaldehyde 12 was found in carrying out the oxidation with selenium dioxide of compound 11 in aqueous dioxane at moderate temperatures [22]. Condensation of compound 12 with hydrazines resulted in corresponding hydrazone compounds 13A1-13A4 [23]. All chemical structures were confirmed by electrospray ionization mass spectrometry and nuclear magnetic resonance spectroscopic analyses.
In vitro antifungal activity was measured by means of the MIC using the serial dilution method in 96-well microtest plates. Test fungal strains were obtained from the ATCC. The MIC determination was performed according to the National Committee for Clinical Laboratory Standards (NCCLS) recommendations with RPMI 1640 (Sigma) buffered with 0.165MMOPS (Sigma) as the test medium [24]. The MIC value was defined as the lowest concentration of test compounds that resulted in a culture with turbidity less than or equal to 80% inhibition when compared with the growth of the control. Test compounds were dissolved in DMSO serially diluted in growth medium. The yeasts were incubated at 35 °C and the dermatophytes at 28 °C Growth MIC was determined at 24 h for Candida species, at 72 h for Cryptococcus neoformans, and at 7 days for filamentous fungi.
3. Results and discussionThe antifungal activity of targeted compounds was listed in Table 1. Most targeted compounds exhibited antifungal activity against the five tested fungi. Compounds 8, 9 and 10 showed broad potency against Candida tropicalis, Trichophyton rubrum and Microsporum gypseum. Compounds 8, 9 and 10 were 2 to 60-folds more potent against C. tropicalis than fluconazole with their MIC80 values in the range of 0.24-7.21 μmol/L. Compound 9A2 showed a better antifungal activity against the 5 tested fungi in vitro than fluconazole, especially against T. rubrum and M. gypseum. On the C. neoformans strain, compounds 8A1, 8A2, 8A4, 8A5, 9A1-9A4 and 13A3 were more active than fluconazole. Compound 5 exhibited weaker antifungal activity than the other compounds against the five tested fungi. Compound 5A2 was active against C. tropicalis at a concentration of 8.72 μmol/L. Compounds 8A and 9A were more active than fluconazole against the 5 tested fungi except Candida albicans. Compound 9A2 was the most potent compound (minimum inhibitory concentration for 80%, MIC80 0.24 μmol/L) against C. tropicalis. Compound 13A showed moderate inhibitory activity against the five tested fungi. However, they were not very sensitive against C. albicans and C. neoformans and only 13A3 showed moderate inhibitory activity (MIC80 = 7.31 μmol/L, 1.82 μmol/L, respectively).

|
Download:
|
| Scheme 1.The synthesis route of the target compounds. Reagents and conditions: (a) 120 °C, reflux, yield 61.6%; (b) alkylamine, N, N'-dicyclohexylcarbodiimide (DCC), dimethylformamide (DMF), 16 h, yield 30%–60%. | |

|
Download:
|
| Scheme 2.The synthesis route of the target compounds. Reagents and conditions: (a) Appropriate aldehyde, diisopropylamine (DIPA), ethanol (EtOH), microwave (MW), 160–170 °C, 1 h, 22%–33%; (b) lithium aluminum hydride (LiAlH4), tetrahydrofuran (THF), ice bath, yield 65%–78%. | |

|
Download:
|
| Scheme 3.The synthesis route of the target compounds. Reagents and conditions: (a) 2-Oxo-propionaldehyde, sodium pyrosulfite (Na2S2O5); (b) selenium dioxide (SeO2), dioxane, reflux; (c) hydrazine, ethanol (EtOH), 60–70 °C. | |
|
|
Table 1 In vitro antifungal activity of the target compoundsa (MIC80, μmol/L). |
When the quinazolinone (compound 5A) was replaced by with a benzopyran (compound 8A or 9A) or a quinoxaline (compound 13A), the antifungal activity increased. Meanwhile, the antifungal activity of compound 8A or 9A was better than that of compound 13A. From the calculated interaction energies, compound 9A bound to CYP51 with the highest value of interaction energy among all compounds tested (Table 2). Compound 5A, with a methyl group on the benzene ring, had potent antifungal activity, while the replacement of the N-methyl group of compound 5A by an ethyl group (compounds 5B) seemed not to be effective. Compounds substituted with a carbonyl group at position 4, such as 8A1-8A5 exhibited lower antifungal activity than those with a hydrogen group, such as 9A1-9A5. Compounds 8A2 and 9A2, containing an alkyl group with 6 carbon atoms, were most effective, suggesting that the length of the alkyl group was an important determinant of antifungal activity. The antifungal activity of compounds 13A3 with three carbon atoms of Nsubstituted alkyl side chain, was higher than that of the other compounds in 13A. Compound 13A4 with an N-isopropyl hydrazine was much lower than compound 13A3 with an Npropyl hydrazine. The SAR analysis above provided a basis for the modification of heterocyclic derivatives in the future.
|
|
Table 2 The calculated interaction energies of the target compounds. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
To explain the results of the assays for antifungal activity, we proposed a binding mode for 9A2 in the active site of CYP51 from C. albicans using the affinity module within the Insight II 2000 software package [25]. As shown in Fig. 3, the benzopyran ring of 9A2 did not coordinate with the heme group, unlike the azoles. Moreover, the benzopyran ring was located in a hydrophobic pocket lined with VAL510, LEU511, TYR118, and HIS377. The alkyl side chains of the compounds were oriented into the pocket, where they formed hydrophobic and van der Waals interactions with GLY309, LEU301, ILE302, TRY132, CYS134, and LEU305. Compound 9A2, with a methoxy group at the position 6 of the benzopyran ring, exhibited reasonable antifungal activity in subsite S4, which suggested that hydrogen-bond donors and acceptors of the position 6 of the molecule influenced its antifungal activity.
4. ConclusionIn response to the need for novel heterocyclic antifungal agents, we designed, synthesized a series of heterocyclic compounds and their antifungal activity was evaluated in vitro. The results indicated that the targeted compounds 8A, 9A and 13A had potent antifungal effects. Compound 9A2 exhibits best antifungal activity against 5 tested fungi in vitro among all compounds, especially against T. rubrum and M. gypseum. The MIC80 value of compound 9A2 against C tropicalis was 0.24 μmol/L, and it was more effective than the other compounds. The SAR analysis provides some clues about how to optimize the structures of nonazole compounds. The novel structures reported here represent a new chemotype that can be exploited for the development of novel heterocyclic antifungal agents.

|
Download:
|
| Fig. 3.The docking model for compound 9A2 and CYP51. | |
{kind=link}
This work was supported by the Shandong Natural Science Foundation (No. ZR2012HQ026) and the Foundation of Sichuan Provincial Health Department (No. 110480).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2015.04.030.
| [1] | J.J. Castón-Osorio, A. Rivero, J. Torre-Cisneros, Epidemiology of invasive fungal infection, Int. J. Antimicrob. Agents 32(2008) S103-S109. |
| [2] | J.N. Sangshetti, F.A.K. Khan, R.S. Chouthe, M.G. Damale, D.B. Shinde, Synthesis, docking and ADMET prediction of novel 5-((5-substituted-1-H-1, 2, 4-triazol-3-yl) methyl)-4, 5, 6, 7-tetrahydrothieno[3, 2-c]pyridine as antifungal agents, , Chin. Chem. Lett. 25(2014) 1033-1038. |
| [3] | H.A. Gallis, R.H. Drew, W.W. Pickard, Amphotericin B:30 years of clinical experience, Clin. Infect. Dis. 12(1990) 308-329. |
| [4] | D.J. Sheehan, C.A. Hitchcock, C.M. Sibley, Current and emerging azole antifungal agents, Clin. Microbiol. Rev. 12(1999) 40-79. |
| [5] | D.W. Denning, Echinocandins:a new class of antifungal, J. Antimicrob. Chemother. 49(2002) 889-891. |
| [6] | I.A. Casalinuovo, P. Di Francesco, E. Garaci, Fluconazole resistance in Candida albicans:a review of mechanisms, Eur. Rev. Med. Pharmacol. 8(2004) 69-77. |
| [7] | H.L. Hoffman, E.J. Ernst, M.E. Klepser, Novel triazole antifungal agents, Expert Opin. Invest. Drugs 9(2000) 593-605. |
| [8] | S.P. Zhu, W.Y. Wang, K. Fang, et al., Design, synthesis and antifungal activity of carbazole derivatives, Chin. Chem. Lett. 25(2014) 229-233. |
| [9] | A. Lupetti, R. Danesi, M. Campa, M.D. Tacca, S. Kelly, Molecular basis of resistance to azole antifungals, Trends Mol. Med. 8(2002) 76-81. |
| [10] | H.T. Ji, W.N. Zhang, Y.J. Zhou, et al., A three-dimensional model of lanosterol 14 a-demethylase of Candida albicans and its interaction with azole antifungals, J. Med. Chem. 43(2000) 2493-2505. |
| [11] | C.Q. Sheng, Z.Y. Miao, H.T. Ji, et al., Three-dimensional model of lanosterol 14 a-demethylase from Cryptococcus neoformans:active-site characterization and insights into azole binding, Antimicrob. Agents Chemother. 53(2009) 3487-3495. |
| [12] | C.Q. Sheng, W.Y. Wang, X.Y. Che, et al., Improved model of lanosterol 14 a-demethylase by ligand-supported homology modeling:validation by virtual screening and azole optimization, Curr. Med. Chem. 5(2010) 390-397. |
| [13] | B. Yao, H.T. Ji, Y.B. Cao, et al., Synthesis and antifungal activities of novel 2-aminotetralin derivatives, J. Med. Chem. 50(2007) 5293-5300. |
| [14] | H. Tang, Y.J. Zhou, Y.W. Li, et al., Design, synthesis and antifungal activities in vitro of novel tetralin compounds, Chin. Chem. Lett. 19(2008) 264-268. |
| [15] | J. Zhu, J.G. Lu, Y.J. Zhou, et al., Design, synthesis, and antifungal activities in vitro of novel tetrahydroisoquinoline compounds based on the structure of lanosterol 14 a-demethylase (CYP51) of fungi, Bioorg. Med. Chem. Lett. 16(2006) 5285-5289. |
| [16] | W.W. Ning, J. Zhu, C.H. Zheng, et al., Fragment-based design of novel quinazolinon derivatives as human acrosin inhibitors, Chem. Biol. Drug Des. 81(2013) 437-441. |
| [17] | C. Pá rká nyi, D.S. Schmidt, Synthesis of 5-chloro-2-methyl-3-(5-methylthiazol-2-yl)-4(3H)-quinazolinone and related compounds with potential biological activity, J Heterocycl. Chem. 37(2000) 725-729. |
| [18] | I.K. Kostakis, A. Elomn, E. Seguin, M. Ianneli, T. Besson, Rapid synthesis of 2, 3-disubstituted-quinazolin-4-ones enhanced by microwave-assisted decomposition of formamide, Tetrahedron Lett. 48(2007) 6609-6613. |
| [19] | M. Fridé n-Saxin, N. Pemberton, K.D.S. Andersson, et al., Synthesis of 2-alkylsubstituted chromone derivatives using microwave irradiation, J. Org. Chem. 74(2009) 2755-2759. |
| [20] | M. Fridé n-Saxin, T. Seifert, M.R. Landergren, et al., Synthesis and evaluation of substituted chroman-4-one and chromone derivatives as sirtuin 2-selective inhibitors, J. Med. Chem. 55(2012) 7104-7113. |
| [21] | N.L. Maidwell, M.R. Rezai, C.A. Roeschlaub, P.G. Sammes, On the development of NAD(P)H-sensitive fluorescent probes, J. Chem. Soc., Perkin Trans. 1(2000) 1541-1546. |
| [22] | S.E. Page, A. Flood, K.C. Gordon, Electron localisation in electrochemically reduced mono- and bi-nuclear rhenium (I) complexes with bridged polypyridyl ligands, J. Chem. Soc., Dalton Trans. 6(2002) 1180-1187. |
| [23] | M.G. Ponizovsky, A.M. Boguslavsky, M.I. Kodess, V.N. Charushin, O.N. Chupakhin, Synthesis of fused quinoxalines, Mendeleev Commun. 12(2002) 68-70. |
| [24] | National Committee for Clinical Laboratory Standards, Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts:Approved standard, Document M27-A2, 2nd ed., National Committee for Clinical Laboratory Standards, Wayne, 2002. |
| [25] | I.I. Insight, Molecular Simulation Inc., 9685 Scranton Road, San Diego, CA 92121-3752, 2000, p. 1999. |