 2015, Vol.26
2015, Vol.26 



Hydroxyapatite (HA), as the main inorganic component of bones and teeth, has been widely used in hard tissue reconstructions, like coatings for artificial hip joints [1], bone cements [2] as well as scaffolds in craniofacial orthopedics [3], thanks to its excellent biocompatibility and bioactivity [4, 5, 6]. However, the intrinsic brittleness of pure HA still hinders its load-bearing applications like in cortical bone substitutions. This has stimulated the development of various HA/polymer nanocomposites whose mechanical properties, in most cases, are still largely inferior to those of natural cortical bones [7, 8, 9]. The main reasons are thought to be the agglomeration of nanoparticles and the lack of strong interactions between the two phases [10].
Surface modification is the key to improve the dispersibility of nanoparticles in polymer matrices. Fore instance, alkyl chains or biodegradable polyester oligmers have been grafted onto HA nanoparticles [11, 12, 13, 14]. These modifications are carried out on previously aggregated HA particles and some aggregation still remains after that. The resultant nanocomposites just show minor improvement in mechanical properties [13, 14]. Recent advances in HA hydrocolloids [15, 16, 17, 18, 19, 20, 21] provide another possibility to prepare highly homogeneous nanocomposites through colloidal chemistry. The HA colloidal particles, stabilized by some amino acids [15], aminoethyl phosphate [16, 17], citrate ions [18], as well as silk fibroin [19], can be directly blended with water soluble polymers like polyvinyl alcohol to make a homogeneous suspension which is then cast and dried to obtain the corresponding nanocomposite. However, this method is not suitable for hydrophobic polymers like polylactide and polycaprolactone which are more widely used in bone grafts. To solve this problem, a new type of amphiphilic stabilizer, dihydrogen phosphateterminated poly(ethylene glycol)s, has been explored to stabilize HA colloid in both water and organic solvents [20, 21]. Based on these findings, a homogeneous HA/polyurethane nanocomposite with 10 wt% of HA in it was generated through a simple solution casting method, with a gain of 75% in tensile strength relative to that of pure polyurethane [9].
Most of the previous surface modifiers can tightly bond onto HA surface, but still lack reactivity with polymer matrices, which may hinder further improvement of mechanical properties of the resultant nanocomposites. We recently synthesized a family of aminoalkyl phosphates [22] that can stabilize HA colloids while generating reactivity for further modifications. Their effects on the structure and properties of HA nanoparticles are reported in this study, as the first step toward new nanocomposites for bone reconstructions.
2. Experimental 2.1. MaterialsA series of ω-aminoalkyl ammonium hydrogen phosphates, H2N-(CH2)n-OPO3HNH4, (AAP-n, with carbon number n ranging from 3 to 6) were synthesized in our lab according to previously described method [22]. Aminoethyl dihydrogen phosphate (AAP-2) was purchased from Sigma-Aldrich (St. Louis, Missouri, USA). Sodium dihydrogen phosphate and anhydrous calcium chloride were from Kelong Factory of Chemical Reagents (Chengdu, Sichuan, China) and served as P and Ca precursor of HA, respectively.
2.2. Synthesis of aminoalkyl phosphates functionalized hydroxyapatites (Cn-HA)
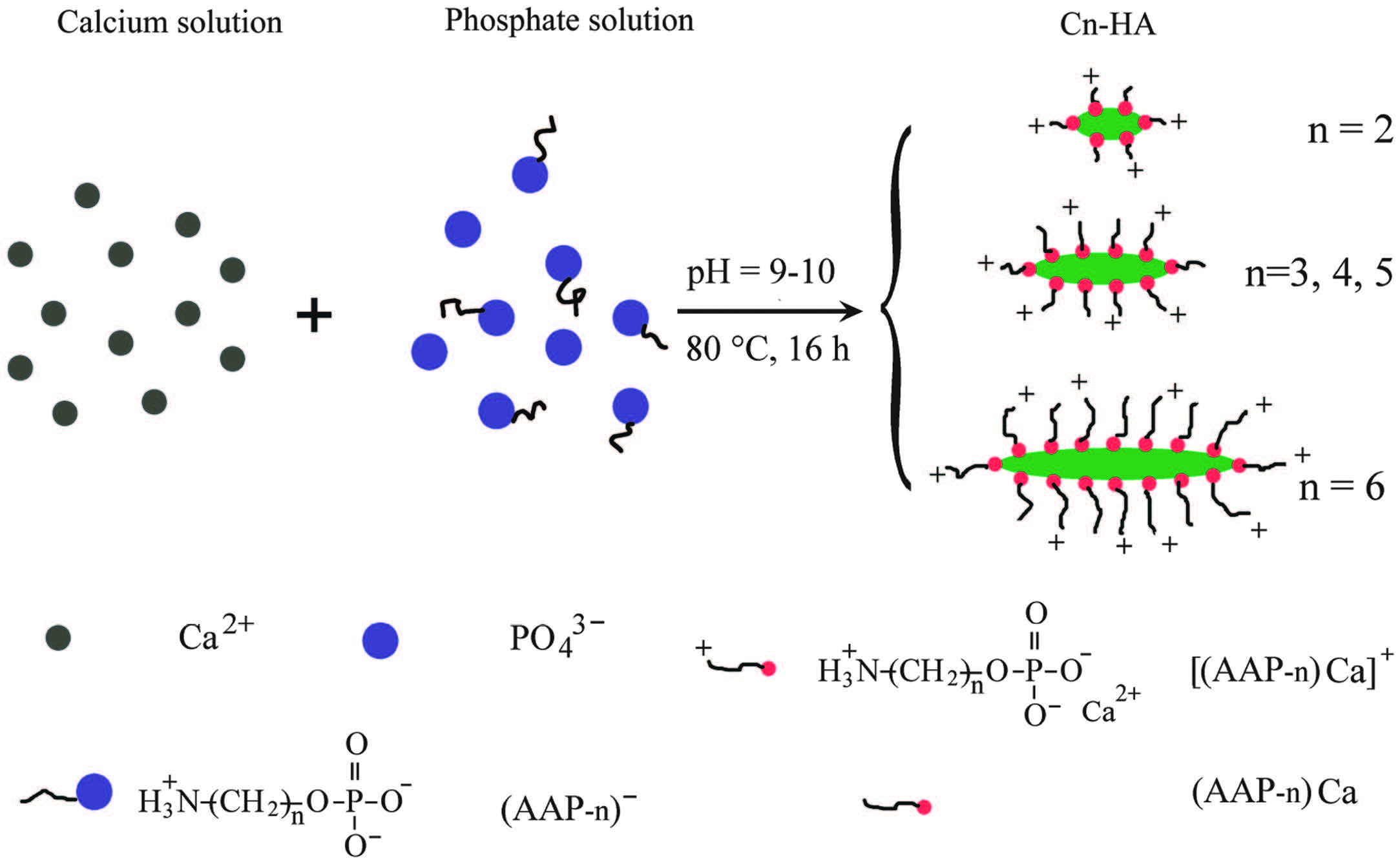
As shown in Scheme 1, each functionalized HA was synthesized by quickly adding a calcium solution into a phosphate solution. The phosphate solution consisted of NaH2PO4 (0.0021 mol) and one type of the surface modifiers AAP-n (n = 2-6, 0.0036 mol) in 15 mL of distilled water. The calcium solution contained 0.006 mol of CaCl2 in the same volume of water. The pH values of both the phosphate solution and the final mixture were adjusted to 9-10, using a solution of 1 mol/L NaOH. The resulting precipitate in the mother liquid was aged at 80 ℃ for 16 h to obtain a light blue hydrocolloid. A control synthesis without any surface modifier only led to HA precipitate. Note that the molar ratios of the organic phosphate from AAP-n and mineral phosphate from NaH2PO4 to Ca2+ ions were kept at 0.6:1 and 0.35:1, respectively, in all the syntheses. The functionalized HAs were designated as Cn-HA, where n represents the carbon number in the structure of the used surface modifier.

|
Download:
|
| Scheme 1.Synthesis of hydroxyapatite nanoparticles functionalized by aminoalkyl phosphates (AAP-n), which serve as both surface modifiers and colloidal stabilizers. Preferential crystal growth along c-axis is observed in C2-HA to C6-HA samples. | |
Five milliliters of each Cn-HA hydrocolloid was kept for colloidal stability observation. The rest was centrifuged at 50, 000 rpm for about 10 min on a Beckman L8-80 M ultracentrifuge (Beckman Coulter, Brea, CA, USA) to obtain a translucent gellike precipitate which was washed three times with water to remove the soluble impurities. All the samples were freeze dried at -50 ℃ and 10 Pa for 24 h and air dried at 60 ℃ for 10 h to obtain powders for various characterizations.
2.3. Characterization 2.3.1. Structures and compositionsThe Fourier transformed infrared (FTIR) spectra of Cn-HA powders were recorded on a Nicolet IR 2000 spectrophotometer (Thermo Nicolet, USA) through KBr disks, with 32 scans between 400 and 4000 cm-1 at a resolution of 4 cm-1. The X-ray diffraction (XRD) patterns were obtained on an X’Pert Pro MPD diffractometer (PANalytical BV, Netherlands) equipped with Cu Kα radiation source operated at 40 kV and 45 mA. The HA (002) reflection was selected to measure the crystallite size (D) according to the Scherrer formula D = 0.89λ/(βcosθ) [23], where λ is the X-ray wavelength (1.54056Å ), β the width in radian at half-maximum height of the diffraction peak, and θ the Bragg angle (12.92°).
The presence of the organic modifiers on Cn-HA particles was tested by thermal gravity analysis (TGA) on a TG 209 F1 thermogravimetric analyzer (Netzsch Group, Germany) from room temperature to 800 ℃, at 10 K/min in a nitrogen flowof 30mL/min.
Total Ca and P contentsweremeasured on VGPQExCell inductive coupled plasma (ICP) emission spectrometer (TJA Corp., USA). The amount of mineral P from PO4 3- groups was determined by spectrophotometry of yellow phosphovanadomolybdate complex (organic phosphate cannot form this complex) according to ISO 11400. In parallel, each samplewas digested with concentrated nitric acid to transformorganic phosphate into inorganic one [24] prior to another determination of total P content using the same spectrophotometry. The difference between the total and mineral P content was the organic content. The C andNcontents in each Cn-HA sample were analyzed using EA3000 elemental analyzer (Euro Vector, Italy).
2.3.2. Colloidal property and stabilityThe particle size and size distribution of each Cn-HA hydrocolloid were measured by photo correlation spectroscopy using Zetasizer 3000 HSa particle analyzer (Malvern Instruments Ltd., Malvern, UK). Zeta potential was also measured on the same instrument. All samples were filtered through 0.45 mm membrane prior to each measurement (n = 3). The dried powders were re-dispersed into various organic solvents by ultrasonication for 4 h (KQ-300DE ultrasonic cleaner, KunshanUltrasonic InstrumentsCo., Ltd, Kunshan, Jiangsu, China). The colloidal stability in water and dimethylformamide (DMF) was recorded by photography using a digital camera.
2.3.3. Morphology of particlesSeveral droplets of the DMF colloid of each sample (1 wt%) was placed onto a copper grid and dried for 24 h at room temperature. Morphology was examined under an H-600IV transmission electron microscope (TEM, Hitachi, Japan).
2.4. Cytotoxicity assayEach Cn-HA powder was extracted with triple-distilled water at 0.1 mg/mL and 37 ℃ for 24 h. The resulting extraction was centrifuged at 3400 rpm through an ultrafiltration tube with a molecular cutoff of 1000 Da (Minipore, USA) to remove any suspending nanoparticles. The centrifuged extractionwas used to dissolve powdered Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies Corporation, Brazil), buffered at pH 7.2 with 5 g/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, Sigma, USA) and 3.7 g/L NaHCO3 (Tianjin Zhiyuan Chemical Reagent Co., Ltd., Tianjin, China). This medium was sterilized by filtration through a 0.22-μm filter (Minipore, USA), and supplementedwith fetal bovine serumat 10% and penicillinstreptomycin solution at 1% (both from Hyclone, Beijing, China) to obtain the sample medium. Normal medium was made the same way except that each sample extraction was replaced with triple-distilled water.
To test cytotoxicity, 100 μL of sample medium was added into some wells of a 96-well plate. One thousand osteoblast-like MG63 cells suspended in 100 mL of sample medium were then added into each of the wells, and incubated at 37 ℃ with 95% air and 5% CO2 for 2, 4 or 6 d (n = 4). The same amount of cells in normal medium and 30% H2O2 were served as negative (blank) and positive controls, respectively. Cell morphology was photographed by an inverted microscope at 2 d. Cell proliferation was determined by the MTT {3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide} assay. After incubation for predetermined time, 20 μL of 5 mg/mL MTT (Sigma, USA) solution was added into each well and incubated for another 4 h in darkness. The medium was discarded and 150 μL of dimethyl sulfoxide was added to dissolve the cell-generated formazan crystals for 10 min. The cell amount was proportional to the optical density of the solution at 570 nm, which was measured on a Multiskan MK3 microplate reader (Thermo Scientific, Shanghai, China).
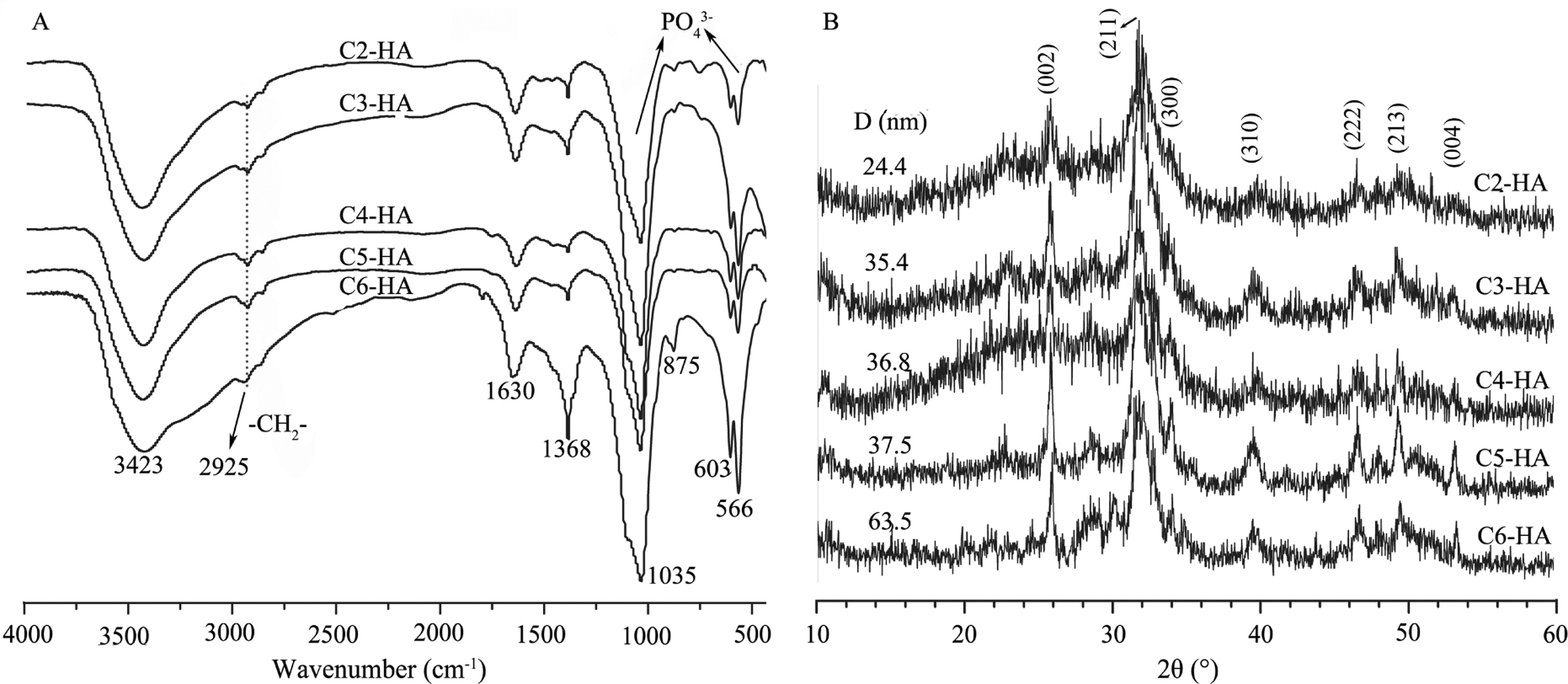
3. Results and discussion 3.1. Chemical structures and compositions of Cn-HAH2N shows the FTIR spectra and XRD patterns of various Cn- HA. Characteristic bands of phosphate stretching at 1035 cm-1 and bending at 603 and 566 cm-1 (H2NA) are found due to the presence of HA [18]. The -CH2- stretching vibration at 2925 cm-1 shows the existence of the surface modifier molecules. The broad adsorption at 3423 cm-1 is related to adsorbed water. The XRD patterns display weak diffraction peaks of HA, like (0 0 2), (2 1 1) and (3 1 0), suggesting low crystallinity of HA in these samples.

|
Download:
|
| Fig. 1. FTIR spectra (A) and XRD patterns (B) of Cn-HA. The -CH2- stretching at 2925 cm-1 shows the presence of organic modifiers. An increase of crystallite size (D) is observed from C2-HA to C6-HA. | |
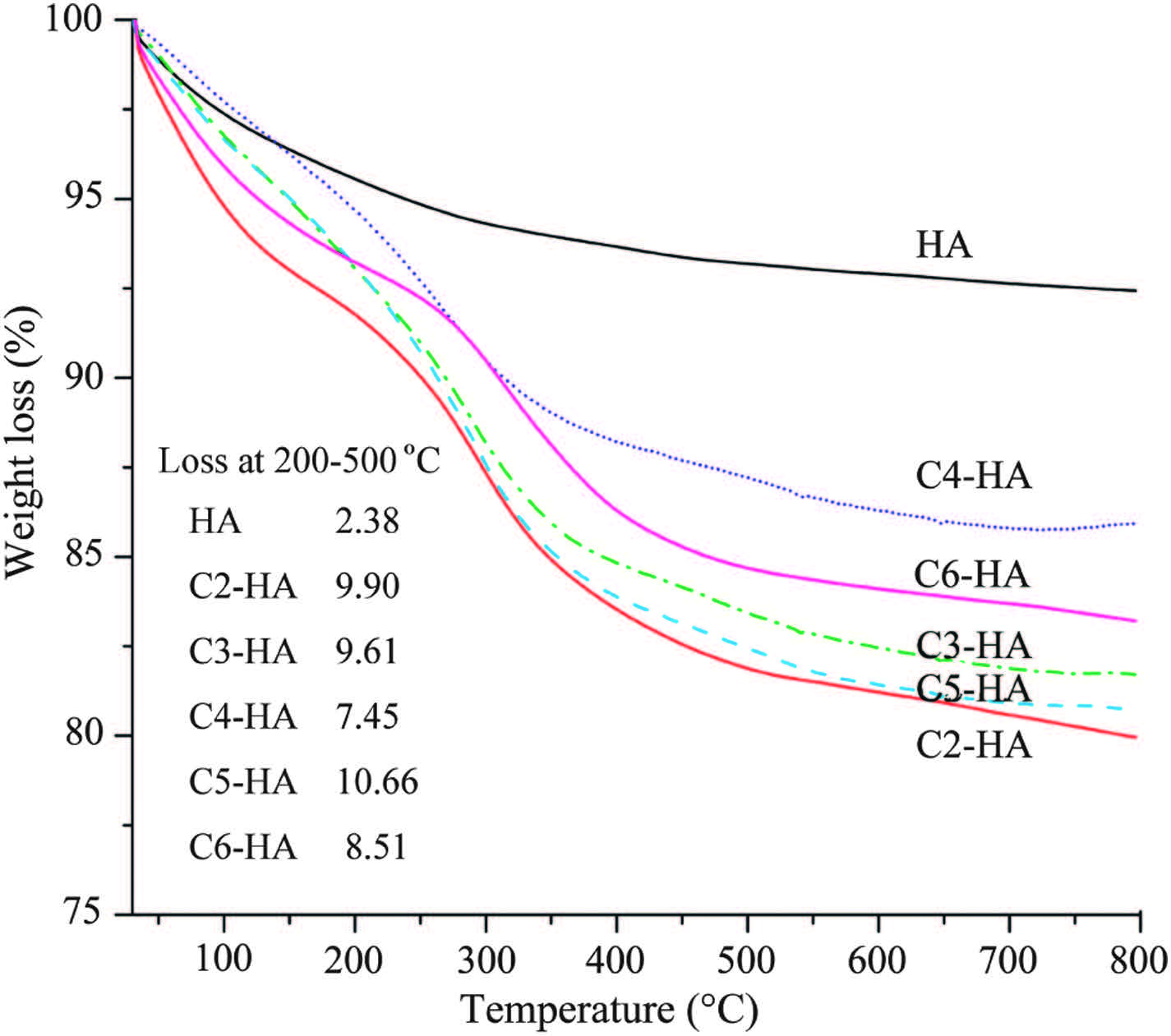
The presence of organic surface modifiers can also be confirmed by TGA (Fig. 2). The obvious weight loss of each Cn-HA in the range of 200-500 ℃, compared with that of HA, can be assigned to the decomposition of the surface modifier. Another weight loss occurred in both Cn-HA and HA at around 100 ℃ is due to the loss of adsorbed water.

|
Download:
|
| Fig. 2.TGA curves of Cn-HA and HA control without surface modification. [(Fig.3)TD$FIG] Fig. 3. Particle size distribution and zeta potential of as-prepared Cn-HA colloids. Table 1 Compositional evaluation of Cn-HA. | |
The composition of each Cn-HA is determined by several techniques (Table 1). Spectrophotometry shows the difference between mineral and total P content. In general, total content is higher than mineral one, showing the presence of organic P fromthe surfacemodifier. Elemental analysis confirms the presence of C and N elements in Cn-HA, both being from the surface modifiers aswell. ICP can not distinguish themineral and organic P, and just gives the total Ca and P content. This P content is comparable to that from spectrophotometry, although some difference has been observed in C2-, C3- and C4-HA, between the two types of measurements.
|
|
Table 1 Compositional evaluation of Cn-HA. |
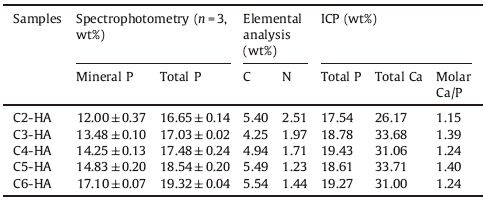
All the above structural and compositional analyses confirm the presence of surface modifiers which must have interacted with the HA in some way. As a matter of fact, the feed ratio of Ca to mineral P is 3.0 in this study, which is much higher than expected value (1.67) from stoichiometric HA. This feed ratio was also used by previous researchers who synthesized HA colloids using AAP-2 as the colloidal stabilizer [16, 17]. At lower feed ratios it is difficult to obtain stable Cn-HA colloids, meaning that excess calcium is needed during the HA synthesis using AAP-n as the stabilizers. The extra calcium ions must be consumed by the stabilizers, forming complex of calcium-(AAP-n), like H2N-(CH2)n-OPO3Ca or [H2N-(CH2)n-OPO3]2Ca, i.e., Ca(AAP-n) or Ca(AAP-n)2. Indeed, in the case of AAP-2 stabilized HA, Chane-Ching et al. proposed a structure model where 4 × 4 cell units of HA in the [ab] plane is surrounded by a peripheral layer consisting of twelve Ca(AAP-2)2 complexes [16]. This layer is not physically mixed with the HA core but intimately interacts with it. The excess Ca ions on one hand interact with outermost AAP-2 ligands through ionic bonds and on another hand coordinate with mineral phosphate groups from internal HA. In this study, the surface modifiers AAP-n are homologues to AAP-2, they can also form a similar outermost layer of Ca-(AAP-n) complex.
We propose that the structure of the outermost complex should be Ca(AAP-n) (Scheme 1), with a Ca/P ratio of 1:1, rather than previously proposed Ca(AAP-n)2 with a Ca/P ratio of 1:2 [16, 17]. Although the latter structure can directly exhibit ammonium groups [+H3N-(CH2)n-] onto the HA particles, these positive charges cannot cause a positive zeta potential since the whole complex is electrically neutral and the outermost ammonium groups are balanced by internal phosphate groups and calcium ions. Moreover, the peripheral cations can adsorb negative ions from local solution and thus the individual HA particles may carry some negative charges. If this is the case, all Cn-HA colloids should display a negative zeta potential. Indeed, all the colloids show positive zeta potentials (Fig. 3B). Therefore, the outermost complex must be H2N-(CH2)n-OPO3Ca, with aminoalkyl groups stretching outwards into the water phase. The pKa2 and pKa3 of AAP-2 is 5.8 and 10.5 respectively [25], meaning that most of the aminoethyl groups are protonated at pH <10.5. Currently, the pKa constants of other AAP-n are not available. However, it can be deduced that their protonation can take place at even higher pH values due to the stronger electron-donating effect of the longer alkylene groups. The pH values of Cn-HA colloids (7-10) are lower than pKa3 of AAP-2 (Fig. 3B), which favor the protonation of the aminoalkyl groups around individualCn-HAparticles, generating electrostatic repulsion among the suspending particles to stabilize the colloids (Scheme 1).

|
Download:
|
| Fig. 3.Particle size distribution and zeta potential of as-prepared Cn-HA colloids. | |
Based on the proposed structure of peripheral complexes [H2N-(CH2)n-OPO3Ca], we can further understand the core-shell structure of Cn-HA particles. The organic P content can be calculated by subtracting mineral P from the total P. The latter is the average of the two values from two different determinationmethods (Table 1). The content of the peripheral organic layer ranges from 16% to 30% (Table 2), calculated based on the respective organic P content. These values are so high that the organic components (at least the Ca(AAP-2) complexwhose content is the highest across all samples) shouldbedetectablebyXRDif they are physicallymixedwith theHA phase. A previous research has clearly shown that Ca(AAP-2) complex prepared in water forms a crystal [26]. However, XRD patterns of the as-prepared Cn-HA all display sole HA diffraction peaks, without any other crystal phase detected. This again confirms the intimate interaction between the peripheral organic layers and the internal HA crystallites, forming a core-shell structure where pure phase of Ca(AAP-n) is absent.
|
|
Table 2 Proposed core and shell compositions of Cn-HA. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The Ca/P molar ratios of the HA cores are lower than standard HA (1.3-1.6 vs. 1.67), suggesting the presence of calcium-deficient HA (Table 2). The internal HA content can be restored based on the measured mineral Ca and P contents and the theoretical HO- content. The total weight percent calculated by combining organic phase and HA phase matches the theoretical value (100%) in samples C3-HA to C6-HA (Table 2). This seems inconsistent with the TGA results showing 2-5 wt% of adsorbed water in Cn-HA powders (Fig. 2). This might be related to the possible readsorption of water before the TGA tests because all the samples were placed in open sample trays waiting to be tested automatically one after another. The insufficient total content of C2-HA (87.7%, Table 2) suggests extra structural water may exist in this sample (see discussion below, in Fig. 6).
3.2. Effects of the surface modifiersThe peripheral Ca(AAP-n) layer can inhibit the growth of HA particles, leading to their narrow particle size distribution in the range of 10-150 nm (Fig. 3A). As previously mentioned, another function of the outermost layer is to exhibit positive charges onto the HA nanoparticles (zeta potential varies from 13 mV to 19 mV) in a weak base environment at pH 7 to 10 (Fig. 3B), to stabilize the HA colloids.
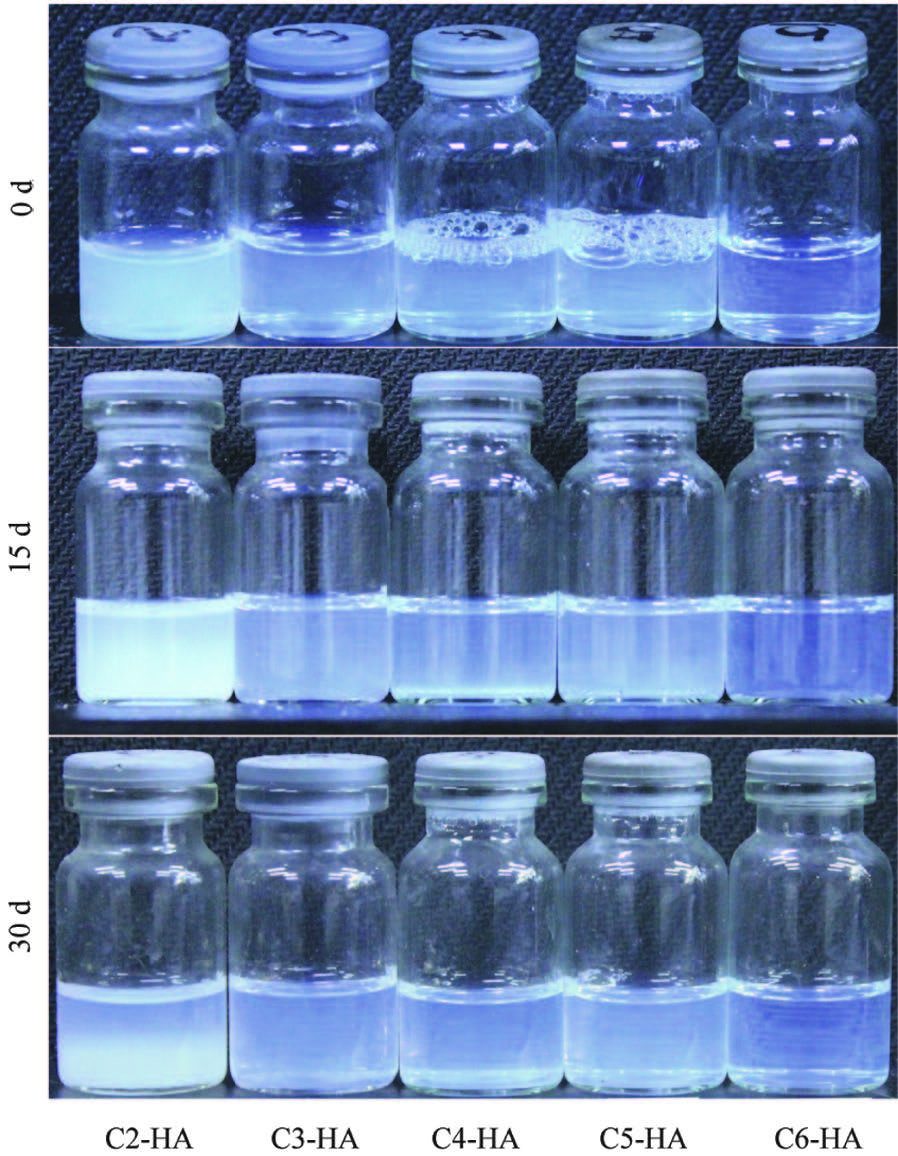
The colloidal stability (Fig. 4) is observed by resting the colloids at room temperature for different period. During the observation period of 30 d, no sedimentation is found for samples C3-HA to C6- HA. The sample C2-HA, however, becomes turbid at 15 d and forms two layers at 30 d (more on this will be discussed later).

|
Download:
|
| Fig. 4.Colloidal stability of as-prepared Cn-HA hydrocolloids. | |
{kind=link}
The freeze-dried Cn-HA powders can be redispersed in organic solvents, like methanol, tetrahydrofuran and dimethylformamide (DMF). However, the stability is far worse than that of the aqueous counterparts. This is shown in Fig. 5 where homogeneous colloid of C3-HA in DMF begins to precipitate at 2 d. Extension the observation to 6 d, the upper solution still displays a light blue colloidal appearance, showing that partial HA particles still suspend in the solution.

|
Download:
|
| Fig. 5.Redispersibility of C3-HA in DMF. | |
{kind=link}
Although the length of aminoalkyl groups (even in C6-HA) is insufficient to stable the colloids in organic solvents, homogeneous colloid-like suspensions can exist for less than 1 d. Considering the reactivity of amino groups on these particles, it is possible to graft organic chains onto these particles to maintain a long-term colloidal stability in organic solvents. Thus various homogeneous HA/polymer nanocomposites could be expected by solution blending and thermally drying.
The carbon number in AAP-n molecules significantly affects the morphology of the corresponding HA nanoparticles. Overall, an increase of aspect ratio is found at the increasing of the carbon number in AAP-n molecules (Fig. 6). For C2-HA, the particles are irregular and shortest (20-30 nm). Spindle morphology is found for samples C3-HA to C5-HA, with similar particle lengths (30- 40 nm) among these samples. C6-HA particles are longest with a rod-like morphology; some are as long as 90 nm. The particle width or diameter, however, is almost the same for all the samples (about 20 nm). This particle size change is perfectly consistent with the crystallite size change measured by half-maximum height of the (0 0 2) diffraction peak (H2NB). This crystallite size, increasing from 24 nm to 64 nm in samples C2-HA to C6-HA, represents the primary particle size along c-axis of HA crystals since (0 0 2) planes are perpendicular to this axis. The increase of crystallite size along c-axis for samples C3-HA to C5-HA can be negligible (H2NB), again consistent with the morphological observation (Fig. 6).

|
Download:
|
| Fig. 6.Morphological change across Cn-HA particles. | |
{kind=link}
Insight into the crystal structure of HA can help us to better understand the morphological change among Cn-HA samples. Perfect HA crystals possess a hexagonal symmetry of P63/m, whose unit cell dimensions are a = b = 9.432Å , c = 6.881Å , and β = 90° [27]. The Ca2+ ions exposed on the side faces parallel to c-axis, (1 0 0) and (0 1 0), form a rectangular lattice of 9.432Å × 6.881Å [28]. These Ca atoms are sparse enough to allow all types of AAP-n (n = 2-6) to bond with, showing similar inhibition to the crystal growth along both a and b directions. This leads to almost the same particle diameters in all Cn-HA. On the other hand, the faces perpendicular to c-axis possess denser Ca2+ ions. For example, the shortest distance between Ca atoms on (001) face can be calculated as 3-0.5a = 5.445Å [27]. This distance allows a fast insertion of relatively small AAP-2 molecules, showing strong inhibition of crystal growth. The middle-sized AAP-n molecules (n = 3, 4 and 5) can bond onto the same crystal face, but at a slower rate than AAP- 2, giving a longer time for crystals to grow along c-axis. For the largest molecules, AAP-6, it is difficult to bond onto (0 0 1) face due to the strongest steric hindrance, showing the least inhibition of crystal growth at c direction and resulting in the longest HA particles. The different steric hindrance of AAP-n molecules can well explain the aspect ratio change across Cn-HA samples (Fig. 6).
An obscure and irregular morphology under TEM is usually related to amorphous calcium phosphate [21], which is a metastable phase toward crystalline HA [21, 29]. Therefore, the increase of image clarity across samples C2-HA to C6-HA is associated with the increase of crystallinity among these samples (Fig. 6). The same increase trend is also found in the XRD patterns where the diffraction peaks become more prominent from C2-HA to C6-HA (H2NB). The near amorphous nature of C2-HA can account for its insufficient total weight percent based on Ca and P contents (Table 2) since amorphous calcium phosphate usually contains 15-20 wt% of structural water in it [21].
The particle size measured by dynamic light scattering (Fig. 3A) seems contradictory to the XRD and TEM results. This implies that some aggregates may exist in the colloidal solution. For example, the zeta potential of C2-HA is the lowest among all samples, suggesting the least stability of this colloid. This is confirmed by the obvious sedimentation in this sample in 30 d (Fig. 4). Note that previous research shows that C2-HA colloids are stable [17]. The instability observed in this study is most likely due to the inappropriate pH value of the solution which is the highest (about 10, Fig. 3B). Their might be some small aggregates of primary particles in this sample which act as individual colloidal particles before precipitating from the colloidal system. Therefore the hydrodynamic particle size measured by dynamic light scattering is the largest despite the smallest primary particle size observed by XRD and TEM. The measured hydrodynamic particle size is sensitive to zeta potential which is related to both pH value of the colloid (Fig. 3B) and the pKa constants of the used surface modifier. Currently only the constants of APP-2 are available [25]. Insight into the change of the hydrodynamic particle size among these samples relies on future research.
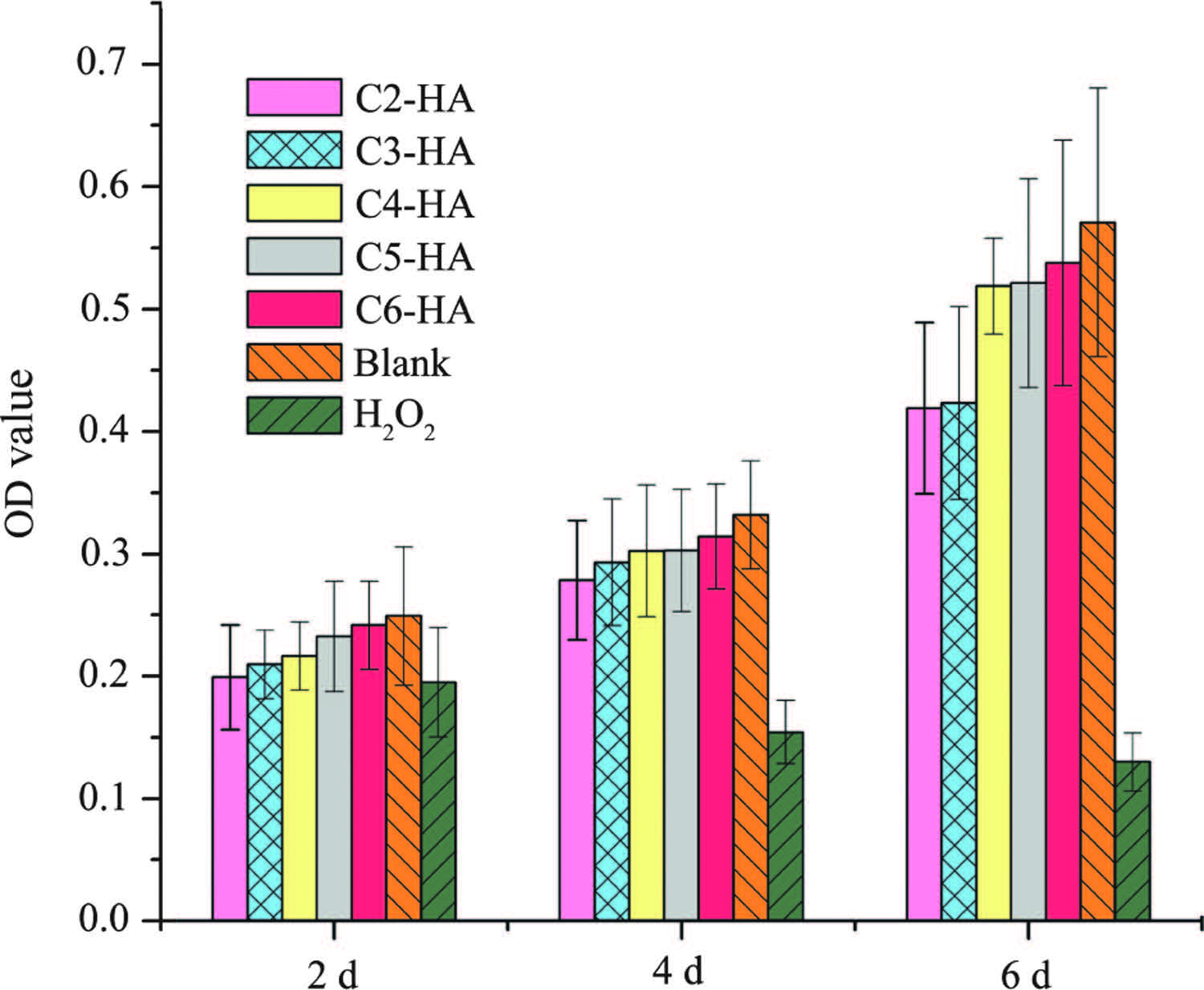
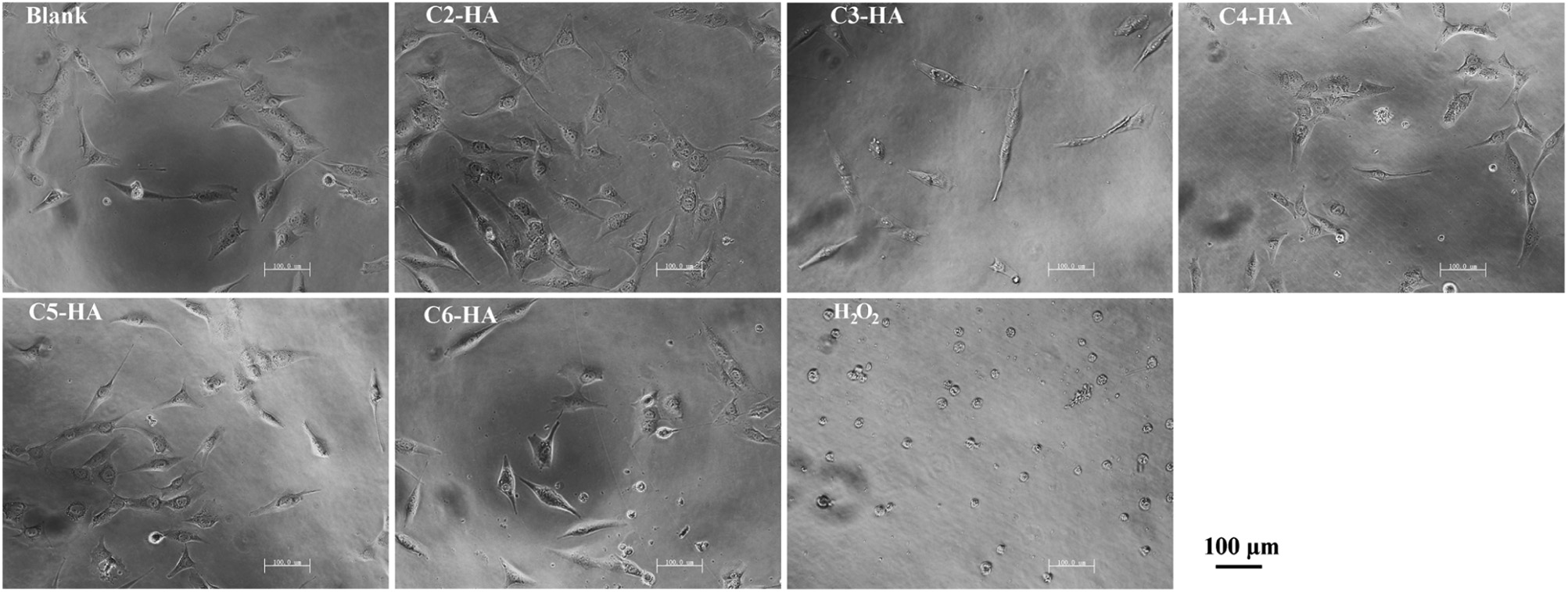
3.3. Cell growth and morphologyAs shown in Fig. 7, the cell viability at 2 d is statistically the same for all test samples and controls. However, at 4 and 6 d, cell viability is significantly lower in 30% H2O2, showing cell toxicity of this positive control. This toxicity is further confirmed by the abnormal round morphology in H2O2 at 2 d (Fig. 8). The cell growth in all Cn-HA extracts is comparable to that in blank control. Except those in H2O2 solution, all cells show a normally elongated morphology (Fig. 8), indicating that no cytotoxicity is associated with Cn-HA.

|
Download:
|
| Fig. 7.Cell growth in Cn-HA extracts. | |
{kind=link}

|
Download:
|
| Fig. 8.Cell morphology after 2-d of culture. | |
{kind=link}
Biocompatibility is essential for any biomaterial. AAP-2 is a natural component found in cell membranes [30], thus it can be regarded as biocompatible. 3-Aminopropane phosphoric acid (acid form of AAP-3) has been reported in a patent to promote fibroblast growth and collagen biosynthesis [31]. Up to now, no more information is available about the biocompatibility of aminoalkyl phosphates and their derivatives.Our cytotoxicity test shows that aminoalkyl phosphates functionalized HAs (Cn-HA) are safe to osteoblast-like MG63 cells, with similar growth rate (Fig. 7) and normal morphology (Fig. 8) as the blank (normal medium) control.
As discussed before, the limited colloidal stability in organic solvents and the reactivity of the peripheral amino groups could provide opportunity to graft organic chains onto Cn-HA particles to make homogeneous HA/polymer nanocomposites. The preliminary results of cell compatibility further solidify that application as bone grafting materials.
4. ConclusionA series of HA hydrocolloids have been synthesized using aminoalkyl phosphates (AAP-n) as both surface modifiers and colloidal stabilizers. Good water dispersibility has been achieved due to the formation of an ionized layer of [(AAP-n)Ca]+ around each HA nanoparticle. This layer preferentially bonds onto the crystal faces parallel to c-axis, allowing preferential crystal growth along this axis. An increase of aspect ratio of HA particles is found at increasing the carbon number in the structure of used AAP-n. The as-prepared HAs demonstrate no cytotoxicity. The amino groups around the HA particles could be used to graft various organic chains to prepare homogeneous HA/polymer nanocomposites as bone grafting materials.
AcknowledgmentsThis work was financially supported by the National Science Foundation of China (No. 50973069) and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (No. 20101561-3-3).
| [1] | R.G.T. Geesink, N.H.M. Hoefnagels, Six-year results of hydroxyapatite-coated total hip replacement, J. Bone Joint Surg. (Br.) 77(1995) 534-547. |
| [2] | D.G. Guo, K.W. Xu, X.Y. Zhao, Y. Han, Development of a strontium-containing hydroxyapatite bone cement, Biomaterials 26(2005) 4073-4083. |
| [3] | H.J. Qiu, J. Yang, P. Kodali, J. Koh, G.A. Ameer, A citric acid-based hydroxyapatite composite for orthopedic implants, Biomaterials 27(2006) 5845-5854. |
| [4] | M.H. Fathi, V. Mortazavi, S.R. Esfahani, Bioactivity evaluation of synthetic nanocrystalline hydroxyapatite, Dent. Res. J. 5(2008) 81-87. |
| [5] | L.W. Lin, K.L. Chow, Y. Leng, Study of hydroxyapatite osteoinductivity with an osteogenic differentiation of mesenchymal stem cells, J. Biomed. Mater. Res. A 89A (2009) 326-335. |
| [6] | W. Zhi, C. Zhang, K. Duan, et al., A novel porous bioceramics scaffold by accumulating hydroxyapatite spherulites for large bone tissue engineering in vivo. II. Construct large volume of bone grafts, J. Biomed. Mater. Res. A 102(2014) 2491-2501. |
| [7] | X.M. Deng, J.Y. Hao, C.S. Wang, Preparation and mechanical properties of nanocomposites of poly (D, L-lactide) with Ca-deficient hydroxyapatite nanocrystals, Biomaterials 22(2001) 2867-2873. |
| [8] | D.A. Wahl, J.T. Czernuszka, Collagen-hydroxyapatite composites for hard tissue repair, Eur. Cells Mater. 11(2006) 43-56. |
| [9] | J.B. Luo, S.X. Qiu, Y.L. Wang, R.H. Lai, X.Y. Xie, Preparation and physicochemical properties of hydroxyapatite/polyurethane nanocomposites, Chin. J. Polym. Sci. 32(2014) 467-475. |
| [10] | M.Šupová, Problem of hydroxyapatite dispersion in polymer matrices:a review, J. Mater. Sci.:Mater. Med. 20(2009) 1201-1213. |
| [11] | H. Tanaka, A. Yasukawa, K. Kandori, T. Ishikawa, Surface modification of calcium hydroxyapatite with hexyl and decyl phosphates, Colloid Surf. A:Physicochem. Eng. Asp. 125(1997) 53-62. |
| [12] | Y.B. Li, W.J. Weng, Surface modification of hydroxyapatite by stearic acid:characterization and in vitro behaviors, J. Mater. Sci.:Mater. Med. 19(2008) 19-25. |
| [13] | Z.K. Hong, P.B. Zhang, C.L. He, et al., Nano-composite of poly (L-lactide) and surface grafted hydroxyapatite:mechanical properties and biocompatibility, Biomaterials 26(2005) 6296-6304. |
| [14] | Y. Wang, J. Dai, Q.C. Zhang, Y. Xiao, M.D. Lang, Improved mechanical properties of hydroxyapatite/poly(ε-caprolactone) scaffolds by surface modification of hydroxyapatite, Appl. Surf. Sci. 256(2010) 6107-6112. |
| [15] | R. Gonzalez-McQuire, J.-Y. Chane-Ching, E. Vignaud, A. Lebugle, S. Mann, Synthesis and characterization of amino acid-functionalized hydroxyapatite nanorods, J. Mater. Chem. 14(2004) 2277-2281. |
| [16] | J.Y. Chane-Ching, A. Lebugle, I. Rousselot, A. Pourpoint, F. Pellé, Colloidal synthesis and characterization of monocrystalline apatite nanophosphors, J. Mater. Chem. 17(2007) 2904-2913. |
| [17] | A. Bouladjine, A. Al-Kattan, P. Dufour, C. Drouet, New advances in nanocrystalline apatite colloids intended for cellular drug delivery, Langmuir25(2009) 12256-12265. |
| [18] | C.C. Li, L.P. Zhao, J.J. Han, et al., Synthesis of citrate-stabilized hydrocolloids of hydroxyapatite through a novel two-stage method:a possible aggregates-breakdown mechanism of colloid formation, J. Colloid Interface Sci. 360(2011) 341-349. |
| [19] | X.W. Huang, X. Liu, S.S. Liu, et al., Biomineralization regulation by nano-sized features in silk fibroin proteins:synthesis of water-dispersible nano-hydroxyapatite, J. Biomed. Mater. Res. B:Appl. Biomater. 102(2014) 1720-1729. |
| [20] | X.Y. Zhou, Y.R. Jiang, C.C. Li, X.Y. Xie, Synthesis of poly (ethylene glycol)-functionalized hydroxyapatite organic colloid intended for nanocomposites, Chin. Chem. Lett. 24(2013) 647-650. |
| [21] | J.B. Luo, S.X. Qiu, X.Y. Zhou, et al., In situ grafting polyethylene glycol chains onto amorphous calcium phosphate nanoparticles to improve the storage stability and organic solvent redispersibility, Colloids Surf. A:Physicochem. Eng. Asp. 444(2014) 81-88. |
| [22] | W.B. Kong, X.Y. Zhou, Y. Yang, X.Y. Xie, A facile synthesis of ω-aminoalkyl ammonium hydrogen phosphates, Chin. Chem. Lett. 23(2012) 923-926. |
| [23] | K.P. Sanosh, M.C. Chu, A. Balakrishnan, T.N. Kim, S.J. Cho, Preparation and characterization of nano-hydroxyapatite powder using sol-gel technique, Bull. Mater. Sci. 32(2009) 465-470. |
| [24] | E.S. Baginski, P.P. Foà, B. Zak, Microdetermination of inorganic phosphate, phospholipids, and totalphosphate in biologic materials, Clin.Chem. 13(1967) 326-332. |
| [25] | J. Christoffersen, M.R. Christoffersen, Kinetics of dissolution of calcium hydroxyapatite:IV. The effect of some biologically important inhibitors, J. Cryst. Growth 53(1981) 42-54. |
| [26] | P. Bissinger, O. Kumberger, A. Schier, Preparation and crystal structures of 2-aminoethyl phosphate complexes of magnesium, calcium, and zinc, Chem. Ber. 124(1991) 509-513. |
| [27] | M.I. Kay, R.A. Young, A.S. Posner, Crystal structure of hydroxyapatite, Nature 204(1964) 1050-1052. |
| [28] | W.G. Jiang, H.H.Pan, Y.R. Cai, et al., Atomicforcemicroscopy revealshydroxyapatitecitrate interfacial structure at the atomic level, Langmuir 24(2008) 12446-12451. |
| [29] | R.H. Lai, P.J. Dong, Y.L. Wang, J.B. Luo, Redispersible and stable amorphous calcium phosphate nanoparticles functionalized by an organic bisphosphate, Chin. Chem. Lett. 25(2014) 295-298. |
| [30] | L. Rothfield, A. Finkelstein, Membrane biochemistry, Annu. Rev. Biochem. 37(1968) 463-495. |
| [31] | O.S. Lee, Y.H. Byon, B.S. Lee, et al., Method for preparing of 3-aminopropane phosphoric acid, US 5723645, 1998. |