 2015, Vol.26
2015, Vol.26 



b Physikalisches Institut and Freiburg Materials Research Center, Albert-Ludwigs-Universität, Freiburg 79104, Germany
Polymer crystallization usually consists of two steps,nucleation and crystal growth. While nucleation plays a key role,it remains so far a poorly understood process. In part,this is because it is still a big challenge to observe experimentally the nucleation event at a molecular level. Thus,theoretical modeling and computer simulation have been employed to reveal the details of nucleation. Lauritzen and Hoffman (LH) proposed the most widely accepted surface nucleation theory of polymer crystallization. There,the free energy for formation of the first crystalline stem (the secondary nucleus) of the polymer chain on the flat growth surface represents the energy barrier for surface nucleation. To allow for growth,the length of this nucleus along the chain direction has to be slightly longer than the minimum required for stability. Under these conditions,the growth rate of the crystal is maximized [1, 2, 3]. In contrast,Sadler and Gilmer (SG) proposed an entropically determined barrier related to the removal of unfavorable chain conformations rather than a barrier due to enthalpy differences [4, 5, 6]. In the SG model lamellar thickening is possible without invoking surface nucleation. The entropic barrier increases with lamellar thickness. The results of the SG model can also explain the correlation of growth rate and lamellar thickness with super-cooling,in accordance with some experimental observations. Along this route,Goldbeck-Wood et al. [7] and Sommer and Reiter [8, 9, 10, 11] considered the molecular kinetics by taking into account interactions between nearest neighbor units. These treatments of molecular kinetics allowed the formation of a rough fold surface of a polymer crystal and agreed in some aspects with experimental results.
However,additional experimental results revealed the existence of meso-phases or intermediate states during crystallization [12, 13]. Although secondary nucleation theory and rate-theory are partly successful on explaining the evolution of the lamellar thickness and the velocity of the growth front,they do not provide a detailed picture of nucleation,neither do they explain the role of intermediate states in crystallization.
Hu et al. have systematically simulated polymer crystallization in the melt and in a confining environment,using dynamic Monte Carlo simulations of systems of lattice chains [14, 15, 16, 17]. They proved that in contrast to the LH model intramolecular crystal nucleation contributes to the nucleation barrier and is responsible for adjacent reentry chain-folding in lamellar crystallites. In their simulations,chain flexibility,interaction between two parallel stems and an energetic barrier for chain sliding were considered. They have revealed a variety of molecular details of the nucleation stage and lamellar growth. Furthermore,Muthukumar et al. took into account the entropy of the connecting loops. Based on Langevin dynamics simulations of a single polymer chain of high molecular weight,they proposed that crystalline lamellae consisting of folded polymers rather than extended chains are thermodynamically stable [18, 19].
Though a large number of works have contributed to the field of polymer crystallization,and particularly nucleation,still many questions have not been answered. For instance,it is not yet clear how anisotropic interactions between chain segments affect polymer nucleation. In this letter,we propose to treat nucleation of polymer crystallization with a model based on microscopic kinetics. We assume that the rate constants of attaching and detaching of a crystalline stem of a polymer chain vary with stem length and the width of the resulting crystalline cluster. We reveal that the formation of secondary nuclei is controlled by multibody interaction parameters.
2. MethodsThe microscopic process of crystallization from a melt can be described well by the cluster-growth scheme,which was adopted by Becker and Do¨ ring [20],Turnbull and Fisher [21]. A series of cascaded elementary processes is applied to simulate the attachment and detachment of a crystalline unit from a crystalline cluster. Becker and Do¨ ring have originally proposed this scheme in 1935 for explaining the gas-liquid phase transition. We now apply this scheme to describe the microscopic kinetics of crystallization. The growth of a polymer crystal results from the processes of attachment and detachment of motifs at the growth surface. Our model considers the basic motif of polymer crystallization to be a short sequence of monomer units. The stem length is determined by the number of crystalline monomer units which they contain. At each site of the crystal,three processes may occur: (1) Extension of a stem by one motif or the initiation of a new stem by attaching a single motif. (2) Simultaneous detachment of a certain number motifs of a crystalline stem. If the stem contains only one motif,it is eliminated once this motif is detached. (3) In cases where the number of attached and detached motifs is balanced,the length of the stem may stay unchanged. The attachment rate constants k+ and detachment rate constants k- determine the rules of evolution of the length of a crystalline stem.
In the original three-dimensional version of kinetic Monte-Carlo simulations performed by Sadler,the attachment rate constant k+ remained unchanged while the detachment rate constant k- was related to the number of bonds broken when a unit is removed [22]. This means that both rates did not change with the length of the crystalline stem. However,for polymer crystallization,the covalent connection between subsequent units along the chain direction and the consequences of these connections have tobe taken into account. Wepropose that along the direction of chain axis,the probability for attachinganadditional crystallineunit toanalready attachedstemof a given number of crystalline units decreases with increasing stem length,i.e.,with increasing number of already crystalline units. This differs fromthe SG model. In the case of small molecules,k+ remains constant,along all the spatial directions,with size of the crystal. Besides,similar to the independence of the attachment rate 1/τA of the crystal size as observed for smallmolecules,in the SGmodel 1/τA is also independent of the stem length,width and orientation of crystalline clusters on the crystalline growth front. Accordingly,the kinetics for attaching of an additional crystalline motif along the chain axis is given by the following expression:
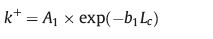
The detachment rate constant depends on the cluster size. Crystallization of small molecules can be considered as the special case where Lc = 1. Considering all the three dimensions of the nucleus of lamellar polymer crystals (lamellar thickness ic along c axis,length ib along b axis and width ia along a axis) of the polymer lamellae,we assume:
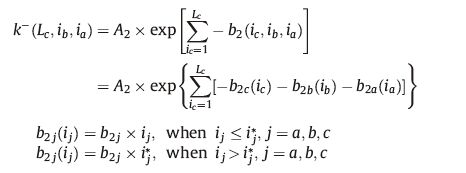
The crystal growth was simulated on the growth face consisting of a cubic lattice of size 100 × 100,with each motif represented by a cube. We use Lc(m,n) to represent the length of stem at position (m,n),where m and n indicates the row and column number, respectively. In a simulation cycle,we calculate the attachment rate k+(m,n) and detachment rate k-(m,n) of each position according to its surroundings,and generate a random number ε(m,n) from 0 to K,where K is the maximum possible range for the detachment rate set in advance. The stem length was allowed to change according to the following principles:
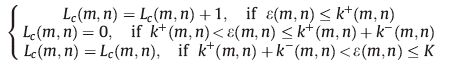
Then,after the time step t = t + 1 the system moved to the next cycle and we repeated the above steps. Thus,we can get the evolution patterns of the whole system.
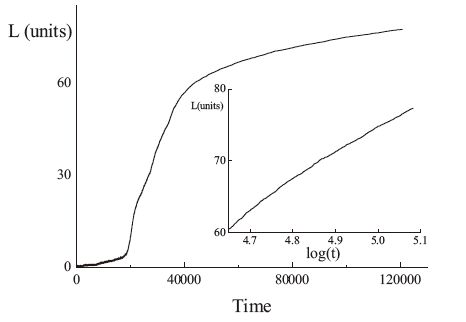
3. ResultsThe average lamellar thickness with time was simulated using a 2-dimensional model,as shown in Fig. 1. As different lengths of stems were allowed in our model,the stem length was not constant but exhibited a distribution in length. At the beginning of the simulation,stems were independently initiated or removed. Repeating this process caused that at some points some stems merged into a cluster,which was large enough to survive in time. At that point,the average thickness of the crystalline lamella started to increase slowly. As we described above,both the attachment and detachment rate constants are related to the size of the crystalline cluster,defined by the mean length of the crystalline stem and its width,i.e.,the number of crystalline stems attached next to each other. Therefore,after an initiation or induction period,the lamellar thickness started to grow,following a logarithmic time dependence:
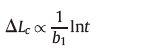

|
Download:
|
| Fig. 1.Variation of chain stem length with time simulated with a 2-dimensional model with b1 = 0.095, b2c = 0.030, b2a = 0.050. The inset redraws this data using a logarithmic time axis. | |
{kind=link}
This logarithmic time dependence is consistent with previous computer simulations [11] and experimental results,for instance, the annealing process of polyethylene (PE) reported by Zhang et al. [23],which showed that the mean thickness of a lamellar crystal increased approximately with the logarithm of annealing time.
The definition of the detachment rate constant k- shows that temperature is not the only factor which influences crystallization. During crystal growth,a competition exists between thickening and broadening of crystalline domains attached at the crystal growth front. Changing the value of b2 allows generating three different crystallization patterns which are dominated by (i) widening,(ii) thickening and (iii) simultaneously thickening and widening of lamellar crystals.
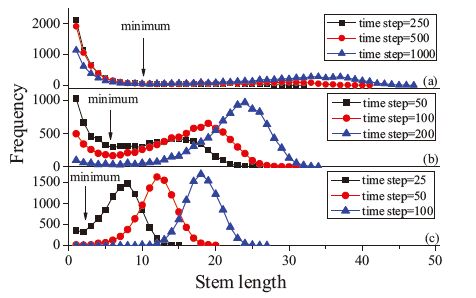
Fig. 2 shows snapshots for these three patterns,obtained under different crystallization conditions and at different times. For b2c and b2a being both small,the initiation of new stems was found to be extremely difficult. Small stems,on the other hand,were not stable and melted. Such stems only became stable when they were about ten units long. Under such conditions,growth of the stem length in the c-direction dominated initially,because chains needed to straighten first in order to reach the minimum length required for stability. Thus,we observed many isolated crystalline stems of a certain length in Fig. 2(a). With the evolution in time, some stems became longer and some of them merged into clusters. Because growth required initiation of long-chain stems,a statistical process,we obtained a quite broad distribution of length of the crystalline stems compared to crystallization patterns observed at early stages of crystallization,as shown in Fig. 3(a). The corresponding process is called lamellar thickening growth.

|
Download:
|
| Fig. 2.Snapshots of two-dimensional simulations with b1 = 0.095 and different b2 values. (a) b2c = 0.030, b2a = 0.050, (b) b2c = 0.030, b2a = 0.095, (c) b2c = 0.095, b2a = 0.150. | |
{kind=link}
However,if we increase b2a and keep b2c unchanged,we can get a different result as revealed in Fig. 2(b). Under these conditions, coalescence of crystalline stems became easier. Given that a long stem was formed initially,short crystalline stems got attached laterally. This process resulted in a dune-shape crystalline cluster. Proceeding in time,illustrating this point,the small cluster became simultaneously wider and thicker. In Fig. 3(b),we can observe that the stem length increased while the distribution in length simultaneously narrowed.
Continuing to increase b2a to 0.150 and b2c to 0.095 in Fig. 2(c), the initiation of new stems became quite easy and yielded a third type of growth pattern,lamellar widening growth. Under these growth conditions,many crystalline stems appeared almost simultaneously across the whole crystalline surface,resembling to some extent a lamellar widening process. These many stems then started almost at the same time to increase in length. The resulting distribution in stem length,shown in Fig. 3(c),also reflects that lamellar thickness increased while the lateral distribution pattern stayed approximately unchanged.
As we discussed above,b2c and b2a both affect the growth of a lamellar crystal,and so does also b2b. From our observations we thus conclude that not only an increase in stem length,but also the lateral attachment of stems,leading to the formation of a cluster, allow to stabilize a secondary nucleus. If the nucleus consists only of a few stems,these stems have to be thicker in order to assure that the nucleus is stable. If the nucleus is wider but consists of shorter stems,it may be equally stable. In the past,only the chain connectivity along the c-direction or the interaction between nearest neighboring crystalline stems were considered when simulating chain adsorption and chain folding. However,multibody interactions between several neighboring stems can be simplified by a mean nearest neighbor interaction. Our results show that there exists a competition between the lamellar thickening and widening processes. Hikosaka et al. have also confirmed that a secondary nucleus tends to grow two-dimensionally after they observed the growth of extended chain crystal (ECC) PE by optical microscopy and the X-ray method [24, 25]. By considering the sliding diffusion of chain molecules with the nucleus,they showed that folded chain crystal (FCC) and ECC are grown under different crystallization conditions. From the stem length distribution diagrams shown in Fig. 3,we can conclude that a minimum stem length exists in each case,corresponding to an energy barrier to be surmounted in surface nucleation.

|
Download:
|
| Fig. 3.Stem length distributions of different growth patterns of two-dimensional simulations with b1 = 0.095; (a) b2c = 0.030, b2a = 0.050, lamellar thickening followed by widening; (b) b2c = 0.030, b2a = 0.095, simultaneous lamellar thickening & widening; (c) b2c = 0.095, b2a = 0.150, lamellar widening followed by thickening. | |
{kind=link}
To simulate secondary nucleation of polymer chains on a flat growth surface we applied three-dimensional modeling of the kinetics. Lauritzen and Hoffman have defined different regimes for describing the transition in growth patterns at different temperatures. At high temperatures,nucleation is difficult while at low temperatures growth is fast and thus dominates the crystallization rate. However,by using a distinctly different model,we obtained similar results in our three-dimensional simulations.
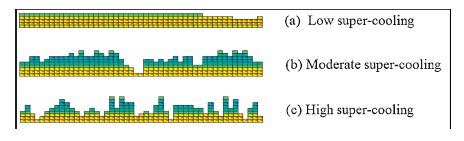
Fig. 4 shows snapshots of crystallization at various supercoolings. At high temperatures,the initiation of new stems at the growth surface was extremely difficult: Thus,the growth front stayed much smoother than at comparatively lower temperatures. At low temperatures,it was easy to stabilize small crystalline stems and thus the growth front was quite rough. We note that, although our model differs from classical nucleation theory,it gives nonetheless,when we vary the values of b2 with temperature, a similar sequence of growth transitions corresponding to the various regimes of the LH-model,which employs classical nucleation theory.

|
Download:
|
| Fig. 4.Growth face patterns at various super-coolings simulated in our model with b1 = 0.095 and (a) low super-cooling, b2c = 0.030, b2a = 0.050, b2b = 0.010; (b) moderate super-cooling, b2c = 0.060, b2a = 0.100, b2b = 0.020; (c) high super-cooling, b2c = 0.090, b2a = 0.150, b2b = 0.030. | |
{kind=link}
In this letter,we have proposed a new model based on the microscopic kinetics of attaching and detaching of crystallizable units of a polymer chain,taking into account long-range multibody interactions. Using appropriate correlation parameters for the attachment and detachment rate constants,we could simulate the evolution of the size and shape of clusters during the nucleation process. Based on two dimensional simulations of the kinetics,we showed that during annealing the average lamellar thickness increased approximately with the logarithm of time,which agrees with experimental observations. Moreover,three distinctly different growth patterns were obtained by changing the correlation parameters: (i) Widening,(ii) thickening and (iii) simultaneously thickening and widening of lamellar crystals,controlled by the corresponding kinetic parameters. Although our model differs from classical nucleation theory,in our three-dimensional simulations of the kinetics of the crystallization process we nonetheless got a similar sequence of regimes with corresponding transitions between these regimes. We expect that in the future, our kinetic model can provide additional insight in the process of polymer crystallization.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (No. 21374054) and the Sino-German Center for Research Promotion. We thank Prof. Wenbing Hu and Prof. Litang Yan for valuable comments.
| [1] | J.I. Lauritzen, J.D. Hoffman, Theory of formation of polymer crystals with folded chains in dilute solution, J. Res. Natl. Bur. Stand. A:Phys. Ch. 64(1960) 73-102. |
| [2] | J.D. Hoffman, J.I. Lauritzen, Crystallization of bulk polymers with chain folding-theory of growth of lamellar spherulites, J. Res. Natl. Bur. Stand. 65(1961) 297-336. |
| [3] | J.I. Lauritzen, J.D. Hoffman, Extension of theory of growth of chain-folded polymer crystals to large undercoolings, J. Appl. Phys. 44(1973) 4340-4352. |
| [4] | D.M. Sadler, G.H. Gilmer, Rate-theory model of polymer crystallization, Phys. Rev. Lett. 56(1986) 2708-2711. |
| [5] | G.H. Gilmer, P. Bennema, Simulation of crystal growth with surface diffusion, J. Appl. Phys. 43(1972) 1347-1360. |
| [6] | D.M. Sadler, G.H. Gilmer, Selection of lamellar thickness in polymer crystal growth:a rate-theory model, Phys. Rev. B 38(1988) 5684-5693. |
| [7] | M.A. Spinner, R.W. Watkins, G. Goldbeck-Wood, Simulation of growth and surface roughening of polymer single crystals, J. Chem. Soc., Faraday Trans. 91(1995) 2587-2592. |
| [8] | G. Reiter, J.U. Sommer, Crystallization of adsorbed polymer monolayers, Phys. Rev. Lett. 80(1998) 3771-3774. |
| [9] | J.U. Sommer, G. Reiter, Polymer crystallization in quasi-two dimensions. II. Kinetic models and computer simulations, J. Chem. Phys. 112(2000) 4384-4393. |
| [10] | J.U. Sommer, G. Reiter, Morphogenesis and nonequilibrium pattern formation in two-dimensional polymer crystallization, Phase Transit. 77(2004) 703-745. |
| [11] | C.F. Luo, J.U. Sommer, Growth pathway and precursor states in single lamellar crystallization:MD simulations, Macromolecules 44(2011) 1523-1529. |
| [12] | T.Y. Cho, W. Stille, G. Strobl, Zero growth temperature and growth kinetics of crystallizing poly(ε-caprolactone), Colloid Polym. Sci. 285(2007) 931-934. |
| [13] | G. Strobl, T.Y. Cho, Growth kinetics of polymer crystals in bulk, Eur. Phys. J. E 23(2007) 55-65. |
| [14] | W.B. Hu, Intramolecular crystal nucleation, in:G. Reiter, G.R. Strobl (Eds.), Progress in Understanding of Polymer Crystallization, Springer, Berlin, 2007, pp. 47-63. |
| [15] | W.B. Hu, Chain folding in polymer melt crystallization studied by dynamic Monte Carlo simulations, J. Chem. Phys. 115(2001) 4395-4401. |
| [16] | Q.Y. Tang, W.B. Hu, Molecular simulation of structural relaxation in ultrathin polymer films, Phys. Chem. Chem. Phys. 15(2013) 20679-20690. |
| [17] | M.Q. Wang, H.H. Gao, L.Y. Zha, et al., Systematic kinetic analysis on monolayer lamellar crystal thickening via chain-sliding diffusion of polymers, Macromolecules 46(2013) 164-171. |
| [18] | M. Muthukumar, Modeling polymer crystallization, in:G. Allegra (Ed.), Interphases and Mesophases in Polymer Crystallization III, Springer-Verlag, Berlin, 2005, pp. 241-274. |
| [19] | M. Muthukumar, P. Welch, Modeling polymer crystallization from solutions, Polymer 41(2000) 8833-8837. |
| [20] | R. Becker, W. Doring, Kinetic treatment of germ formation in supersaturated vapour, Ann. Phys. 24(1935) 719-752. |
| [21] | D. Turnbull, J.C. Fisher, Rate of nucleation in condensed systems, J. Chem. Phys. 17(1949) 71-73. |
| [22] | D.M. Sadler, G.H. Gilmer, A model for chain folding in polymer crystals:rough growth faces are consistent with the observed growth rates, Polymer 25(1984) 1446-1452. |
| [23] | B. Zhang, J.B. Chen, H. Zhang, et al., Annealing-induced periodic patterns in solution grown polymer single crystals, RSC Adv. 5(2015) 12974-12980. |
| [24] | M. Hikosaka, Unified theory of nucleation of folded-chain crystals and extendedchain crystals of linear-chain polymers, Polymer 28(1987) 1257-1264. |
| [25] | M. Hikosaka, K. Watanabe, K. Okada, S. Yamazaki, Topological mechanism of polymer nucleation and growth-the role of chain sliding diffusion and entanglement, Adv. Polym. Sci. 191(2005) 137-186. |