 2015, Vol.26
2015, Vol.26 



b Thermo Fisher Scientific(China)Co., Ltd., Beijing 100080, China
Per- or polyfluoroalkyl substances (PFASs) have been widely used in the textile, paper products, pesticides, carpets, leather, floor polish, firefighting foams, and other industrial and civil applications due to their excellent thermal stability, high surface activities, and hydrophobic/oleophobic properties [1]. The application of PFASs leads to environmental pollution throughout their life cycle including production, transportation, use, and disposal [2]. Thus, the contamination of PFASs in the environment might cause severe toxicity to wildlife and human [3, 4, 5]. In recent years, PFASs have emerged as contaminants of concern because of their broad distribution and persistence in the environment. Given that PFASs are extensively present and very persistent in the environment, and potentially harmful to the environment, wildlife, and human, more and more international organizations have limited and regulated their use and production [6]. For example, World Organization for Economic Cooperation and Development (OECD) has classified perfluorooctane sulfonate (PFOS) as a persistent, bioaccumulative and toxic substance (Persistent Bioaccumulative Toxic, PBT) in 2002 annual risk assessment report [7]. Consequently, PFOS, its salts, and perfluorooctane sulfonyl fluoride were added to the list of Appendix B in the Stockholm Convention on Persistent Organic Pollutants (POPs) in 2009 [8]. Accordingly, their worldwide production and usage will be subjected to restrictions and norms. Nonetheless, the production and use of PFOS and other worldwide perfluoroalkyl substances are still continuing because of the difficulty to discover the ideal replacement for PFOS or the higher cost of alternatives. Therefore, their concentrations would continue to increase or remain high levels in the environment and human samples for many years to come [9, 10].
The premise for the examination of pollution levels, environmental behavior and toxicity of PFASs is a highly sensitive and accurate analytical method. Since the content of PFASs in environmental media is very low, mostly in ng/kg level, analytical method with direct detection requires highly sensitive analytical instruments. Currently, the determination of PFASs requires the pre-treatment of water, soil and biological samples including solvent extraction or solid phase extraction, then concentration and purification, followed by liquid chromatography-tandem triple quadrupole MS detection [11, 12]. Indeed the off-line solid phase extraction coupled to liquid chromatography-mass spectrometry detection (SPE-HPLC-MS/MS) method has become mainstream in the analysis of PFASs. However, the current offline analytical method has some drawbacks and challenges in the analysis of environmental water samples from remote areas, such as Antarctic and Arctic areas. For example, the off-line procedure tends to cause complex cross-contamination. The off-line SPE procedure is not a full sample loading method, and when it is used, only a small fraction of the extracts (typically, 5-10 mL in 1-2 mL) is injected to HPLC-MS/MS system for analysis, which definitely reduces the enrichment and sensitivity greatly. Environmental water samples with extremely low concentration usually require pre-concentration of several or even dozens liters of samples prior to HPLC-MS/MS analysis. Thus, it is very challenging to prepare samples under certain circumstances, especially in extreme environments and remote areas. Obviously, on-line solid phase extraction techniques may overcome shortcomings of the off-line liquid chromatography-mass spectrometry detection technology. Solid phase extraction technology achieves automatic sample concentration, purification, and analysis. This technology exhibits many advantages such as excellent reproducibility, simple pretreatment, and higher accuracy by avoiding human errors that are introduced in the off-line solid phase extraction procedures. More importantly, on-line solid phase extraction is a full loading method, in which all extracts are transferred to the HPLC-MS/MS for the analysis without any loss. Consequently, this technique could considerably enrich analyte and significantly improve the sensitivity using a limited volume of sample, which is important for the analysis of trace components in extremely valuable samples in remote areas, such as Antarctic and Arctic areas. Therefore, the on-line extraction technique has been rapidly developed and widely used in the analysis of a variety of contaminants in environmental and biological samples since it was invented [13, 14, 15]. However, to the best of our knowledge, the analysis of perfluoroalkyl substances using on-line solid-phase extraction technique has been rarely reported.
In this work, we developed a method for the analysis of PFASs in environmental water samples using on-line solid phase extraction- liquid chromatography-tandem mass spectrometry (On line- SPE-HPLC-MS/MS). After simple filtration, the environment water samples can be analyzed directly using this on-line hyphenation method, which integrates sample collection, chromatographic separation and mass spectrometric detection into one process. This method shows numbers of advantages such as low sample consumption, high sensitivity, high degree of automation, and minimum human impact. We have obtained satisfactory results by utilizing this method in the analysis of PFASs in Antarctic ice core samples.
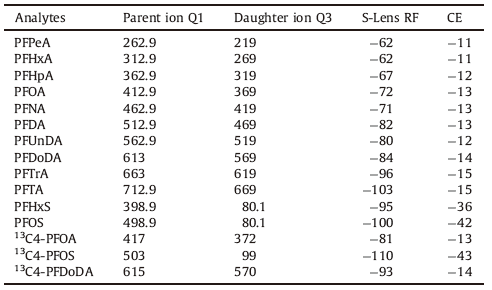
2. ExperimentalAll native PFASs, including perfluoropentanoic acid (PFPeA), perfluorohexanoic acid (PFHxA), perfluoroheptanoic acid (PFHpA), perfluorooctanoic acid (PFOA), perfluorononanoic acid (PFNA), perfluorodecanoic acid (PFDA), perfuoroundecanoic acid (PFUnDA), perfluorododecanoic acid (PFDoDA), perfluorotridecanoic acid (PFTrDA), perfluorotetradecanoic acid (PFTeDA), perfluorohexanesulfonate (PFHxS), PFOS, and mass-labeled PFASs (13C4-PFOA, and 13C2-PFDoDA, 13C4-PFOS) were purchased from Wellington laboratories. Methanol (HPLC grade) was obtained from Fisher Scientific (Pittsburgh, PA, USA). Ammonium acetate (NH4OAc, >97%), ammonium hydroxide (v/v, 50%, HPLC grade), acetic acid (>99.8%, HPLC grade), formic acid (>98%, HPLC grade) were obtained from Alfa Aesar (Ward Hill, MA, USA). Water (>18.2 MV cm) used in the present study was purified on a Milli-Q Advantage A10 system (Millipore, USA).
On-line solid phase extraction system consists of dual-gradient rapid separation pump DGLC-3600RS, column oven TCC-3200, and autosampler WPS-3000 TLS. The entire analysis process is effectively controlled by chromatography workstation Chromeleon 6.70. Five different types of commercially available on-line solid phase extraction columns, C18 (C18 bonded to 50 mm silica, 4.3 mm × 10 mm), SolEx RSLC HRP (hydrophilic divinylbenzene polymer particle with 12-14 mm, 2.1 mm × 20 mm), TurboFlow Cyclone (60 mmpolymer, 1.0 mm × 50 mm), TurboFlow Cyclone-P (60 mm polymer with modified surface, 1.0 mm × 50 mm), and TurboFlow Cyclone MAX (mixed-phase anion exchanger bonded to 60 mm polymer, 1.0 mm × 50 mm), were investigated to assess their SPE performances. On the basis of their SPE performances, SolEx RSLC HRP was selected as the on-line solid phase extraction column. Acclaim 120 C18 (2.1 mm × 100 mm, 2.2 mm, Thermo Fisher Scientific) was used as the separation column. The ternary mobile phase consisted of methanol (A), 5 mmol/L ammonium acetate in water (B), and deionized water (C). The flow rate was 0.25 mL/min. The water samples were injected into a triple quadrupole mass spectrometer TSQ Access MAX for detection after the pretreatment using automatic concentration and separation. The mass spectrometer was operated in the negative ion MS/MS mode (-ESI) using Multiple Reaction Monitoring (MRM) mode. The following were MS-related parameters, Sheath gas: 40 units; Aux gas: 30 units; Source voltage: -32 kV; Vaporizer temperature: 350 8C; Capillary temperature: 350 8C; Scan time: 0.01 s. Other parameters and analyzed compounds were shown in Table 1.
|
|
Table 1 Experimental condition of electrospray tandem mass spectrometry for PFASs. |
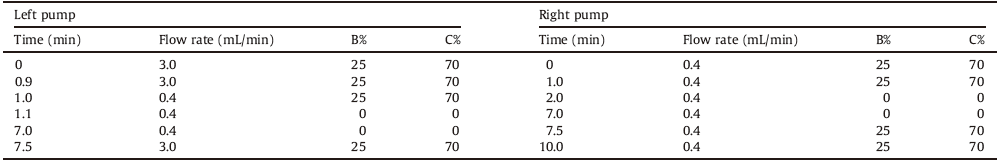
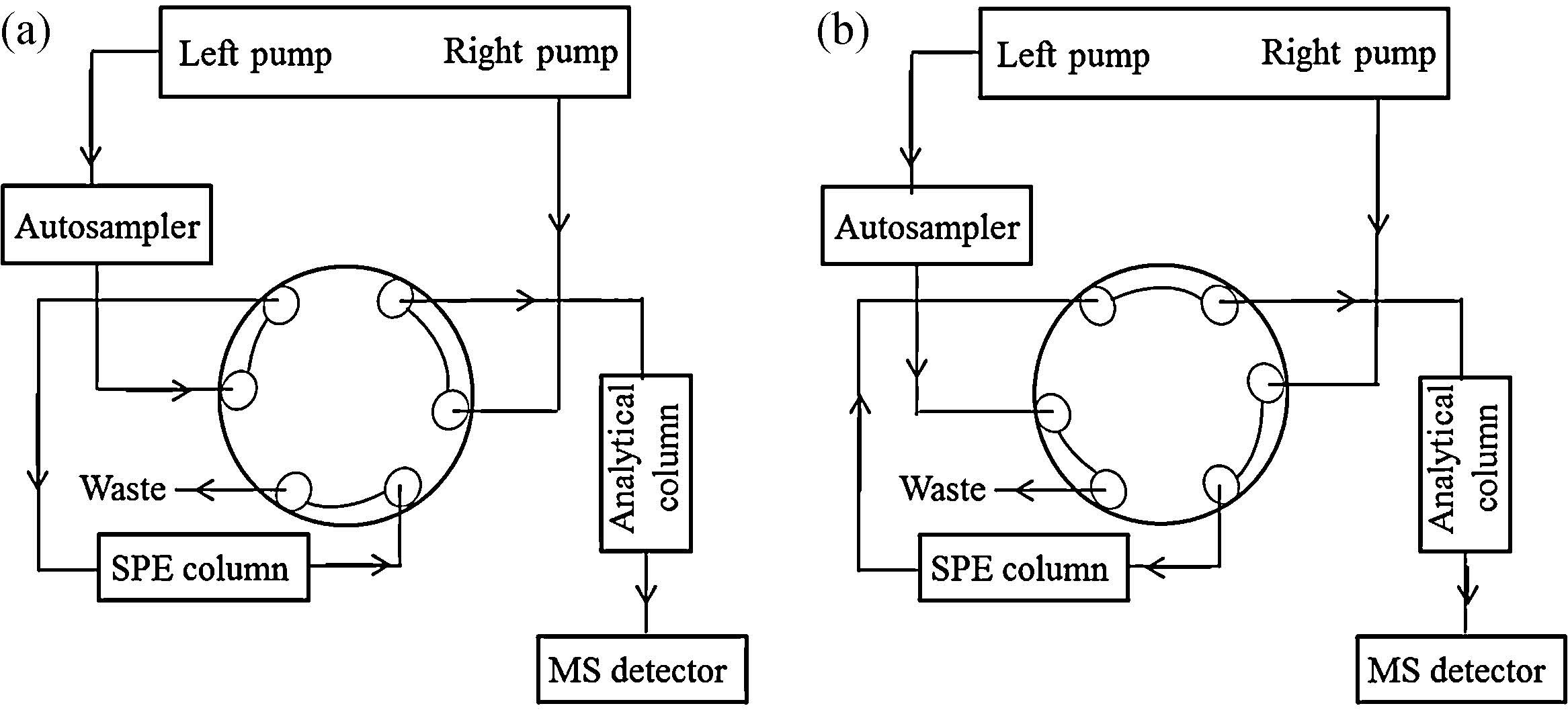
The principle of on-line solid phase extraction was shown in Fig. 1. Firstly, a large volume of sample (2.5 mL in this study) was directly injected into an on-line solid phase extraction column using the auto-sampler. Then impurities on extraction column were flushed into the waste by the left pump in dual gradient liquid chromatography with a pre-set solvent cleaning program. After the completion of cleaning, the extraction column was switched to the analytic column via a valve. Subsequently, the analytes in the extraction column were eluted and transferred into the analytical column for separation by the right pump of HPLC with the chromatographic mobile phase. Finally, the separated analytes were detected by the mass spectrometer. The on-line extraction conditions and procedures were shown in Table 2.

|
Download:
|
| Fig. 1.Instrumental configuration of the on-line SPE. (a) Sample was loaded to SPE column (valve: load position), (b) sample was transferred to analytical column (valve: inject position). | |
|
|
Table 2 The on-line extraction conditions and procedure. |

|
Download:
|
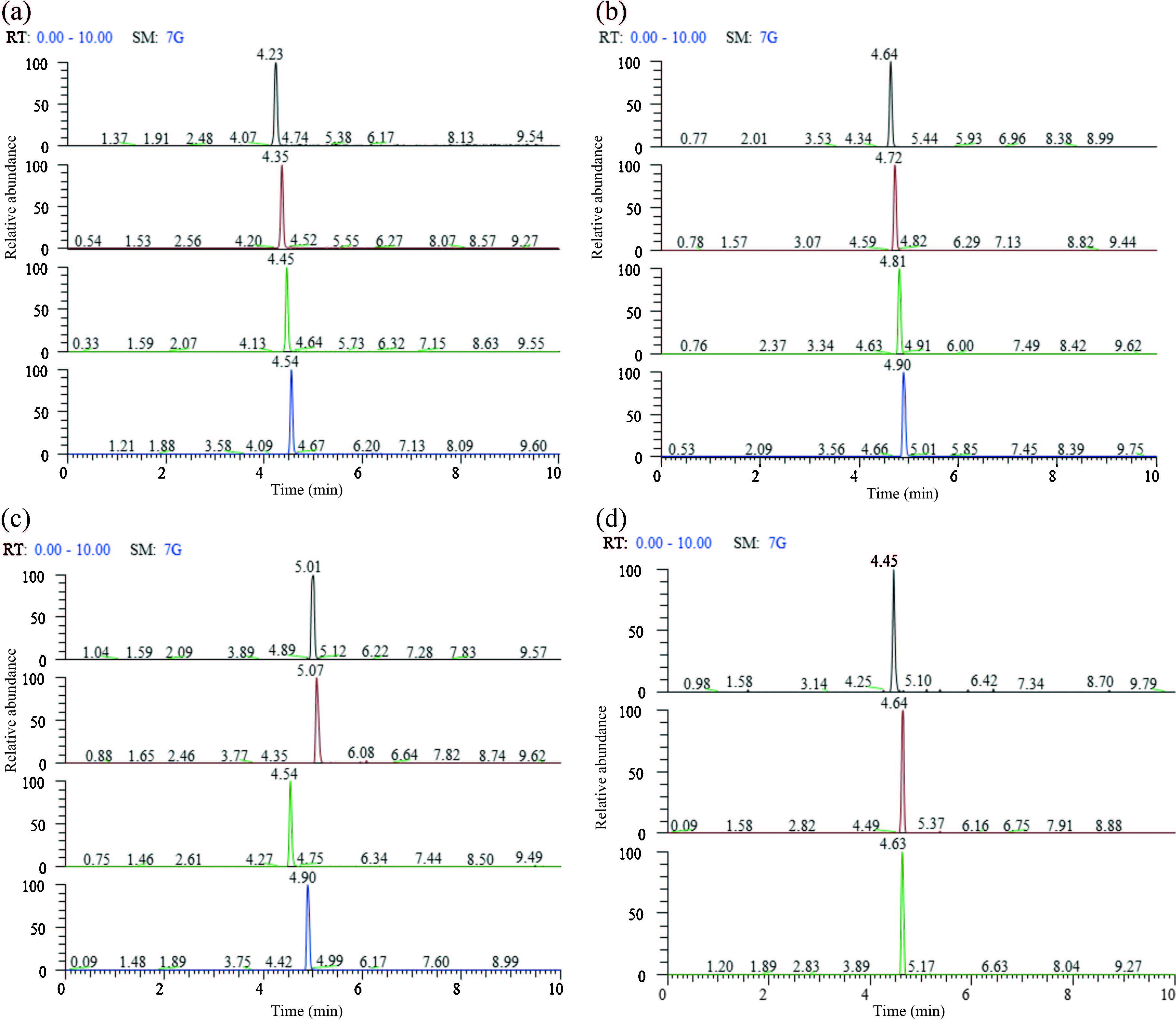
| Fig. 2.The chromatogram of PFASs with the concentration of ammonium acetate at 0.2 mmol/L (a), 1 mmol/L (b), 5 mmol/L (c), and 10 mmol/L (d) in mobile phase. | |
3. Results and discussion
Ammonium acetate is commonly used as the buffer salt in mobile phase to improve the separation, peak shape, and response sensitivity of chromatography. Herein we firstly examined the effects of four different concentrations (0.2, 1.0, 5.0, and 10.0 mmol/L) of ammonium acetate in mobile phase on the separation, peak shape, and sensitivity of PFASs. As shown in Fig. 2, increasing the concentration of ammonium acetate from 0.2 mmol/L to 5.0 mmol/L resulted in sharper, higher and more symmetrical peaks and significantly improved resolution and sensitivity of PFASs. However, when the concentration of ammonium acetate was increased from 5 mmol/L to 10 mmol/L, the detection sensitivity of PFASs did not change evidently. Therefore, considering the peak shape, sensitivity, impact of ammonium acetate on MS, we selected 5.0 mmol/L ammonium acetate as a buffer solution for the mobile phase.
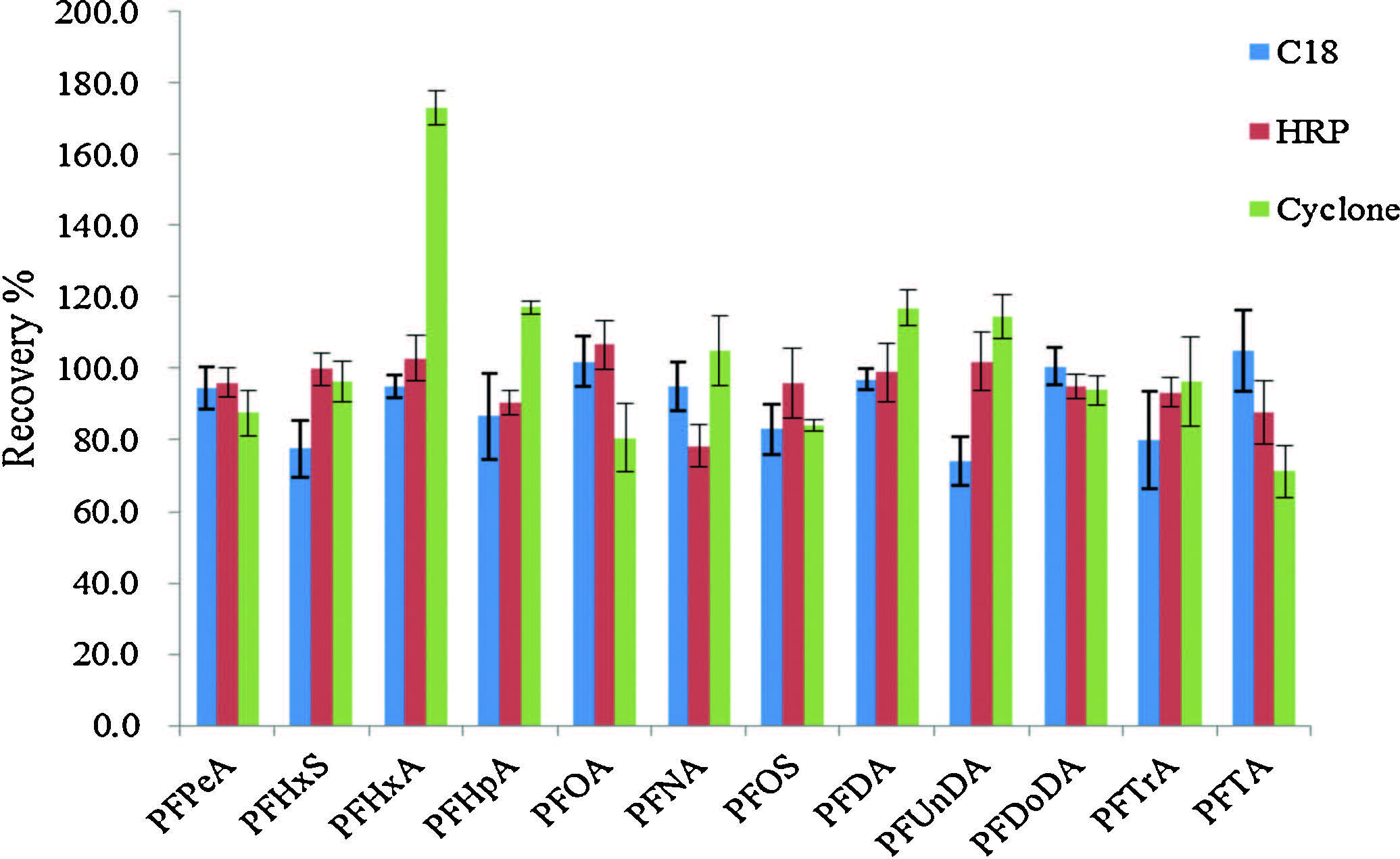
The enrichment efficiency of PFASs from the water sample is a critical factor to evaluate and select the on-line SPE column. Therefore, we rationally selected five different types of commercially available on-line solid phase extraction columns, C18, HRP, TurboFlow Cyclone, TurboFlow Cyclone-P, and TurboFlow Cyclone MAX to investigate their extraction efficiencies for PFASs. The results indicated that the C18, HRP and Cyclone columns had good retention for all PFASs. However, Cyclone-P and Cyclone MAX columns exhibited poor retention for short chains (C4-C7) and medium chains (C8-C9) PFASs, respectively. The performance differences of these columns could be attributed to the polarity differences of packing materials. The packing materials of C18, HRP and Cyclone columns are hydrophobic, while those of Cyclone-P and Cyclone MAX are polymers with some polar modifications, which are less hydrophobic than those of C18, HRP and Cyclone columns. According to literature reports [11], most of PFASs are retained mainly via hydrophobic interactions, which could explain the differences of retention performance of these columns. Consequently, Cyclone-P and Cyclone MAX columns did not meet the requirements of the extraction and enrichment. Next we further evaluated the extraction efficiencies of C18, HRP and Cyclone columns for PFASs. The results showed that the extraction efficiency of Cyclone column was slightly lower than 71% for PFTA, but was more than 150% for PFHxA. C18 and HRP columns can extract all analyzed PFASs with extraction efficiencies in the range of 75%-110% (Fig. 3). Considering the lifetime of column, activation and elution conditions, eventually we selected HRP as the on-line SPE column for this study.

|
Download:
|
| Fig. 3.Extraction efficiency of PFASs in C18, HRP and Cyclone. | |
Having selected SPE column, we next optimized the switching time of the sampling valve that was fitted with SPE column to ensure all PFASs were transferred into the separation column. The results showed that the response of PFASs stopped increasing after 5 min interval, indicating all PFASs were transferred into the separation column in 5 min. However, the valve switching process might influence the separation or peak shape. To avoid the potential interference, we set the time to switch sampling valve at 6 min interval. In addition, we also examined the sample loading rate and volume because the sample loading rate may affect the extraction efficiency of the analytes and the loading volume may directly affect the detection sensitivity of the analytes. Firstly, we investigated the effect of sample loading rate on the extraction efficiency of PFASs. Comparing the loading rates of 1.0 mL/min, 2.0 mL/min, 3.0 mL/min, and 4.0 mL/min, we found that there was no significant variation in the response of PFASs when the rate increased from 1.0 mL/min to 3.0 mL/min. However, the responses of PFOS, PFHxA and PFHpA declined in different percentages when the loading rate was increased to 4.0 mL/min, suggesting there were adverse effects on the extraction efficiency of certain compounds when the loading rate exceeded 3.0 mL/min. Therefore, we selected 3.0 mL/min as sample loading rate. Subsequently, we examined the effects of different loading volumes (1.0 mL, 1.5 mL, 2.0 mL, and 2.5 mL) on the extraction efficiency. The results showed that responses of all PFASs increased linearly with the increasing loading volumes at the fixed loading rate (3.0 mL/min), indicating that all PFASs were well retained in the SPE column for all the selected loading volumes. Finally, considering the injection time and response intensity, we selected 2.5 mL as the sample loading volume.
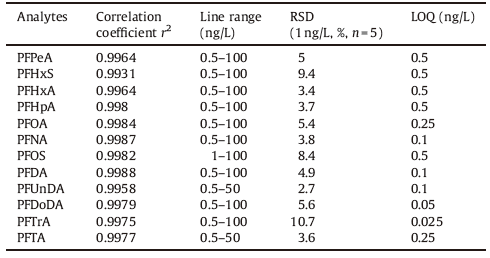
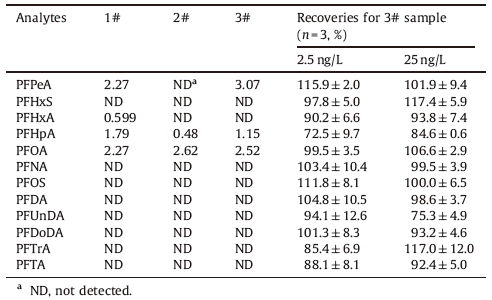
The optimal separation and extraction were achieved for all 12 different PFASs by using the developed optimal analytical conditions. Fig. 4 is the chromatogram of a mixed standard solution at the concentration of 2.5 ng/L with a loading volume of 2.5 mL. Furthermore, we evaluated the linear range, reproducibility, sensitivity, and other related analysis performances of this analytical method. The results indicated that this method displayed good linearity (r2 > 0.99) for all 12 PFASs over the concentration range of 0.5-100 ng/L. The limits of quantitation for samples were in a range of 0.025-0.5 ng/L and the relative standard deviations (RSD) of five consecutive analysis were less than 10% for 1 ng/L standard solution (as shown in Table 3). It is noteworthy that the LOQs obtained using only 2.5 mL of water sample are comparable to or lower than those obtained using conventional off-line SPE-HPLC-MS/MS methods that consume several hundred milliliters of water sample [11, 16]. Finally, we applied this method to the determination of PFASs in Antarctic ice core samples, which were collected from the summit of a Dome (808220 S, 778220 E) in the 2004/2005 austral summer [17]. It turned out that both PFOA and PFHpA were detected in all three samples, PFPeA was detected in two samples, and PFHxA was only detected in one sample. In addition, the recoveries of analytes were between 73% and 117% (as shown in Table 4) when samples were spiked with standards at the concentrations of 2.5 ng/L and 25 ng/L. This result demonstrated that the developed method could be used for the determination of PFASs in the Antarctic ice core samples.

|
Download:
|
| Fig. 4.The chromatogram of 25 ng/L PFASs with 2.5 mL direct injection. | |
|
|
Table 3 Linearity, correlation coefficients and limits of quantification. |
|
|
Table 4 Concentrations of PFASs found in the Antarctic ice core samples and recoveries for the samples spiked with 2.5 ng/L or 25 ng/L PFASs. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In conclusion, we have developed an on-line solid phase extraction-liquid chromatography-tandem mass spectrometry method to analyze PFASs in environmental water samples. Since this method only needs very simple pre-treatment, the river water or tap water samples can be analyzed directly after simple filtration. Most importantly, the outstanding advantage of this method is the full loading of a sample. Therefore, this method could considerably enrich analytes and significantly improve the sensitivity using a limited volume of sample. This method is especially suitable for the analysis of trace components in extremely valuable samples in remote areas, such as Antarctic and Arctic areas. In addition, this method also addressed some issues of off-line analysis techniques, such as complicated procedures, poor detection sensitivity, and cross-contamination. Since this method requires less manpower and resource, it is more economic compared with the off-line technique. Taken together, we believe that the on-line SPE LC-MS/MS method will play more important roles in the analysis of trace amount of PFASs in environmental samples and their environmental behavior studies. Acknowledgments
This work was jointly supported by the National Natural Science Foundation of China (Nos. 21377145, 21321004) and the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB01020300).
| [1] | S. Posner, Perfluorinated compounds:occurrence and uses in products, in:T.P. Knepper, F.T. Lange (Eds.), Polyfluorinated Chemicals and Transformation Products, Springer, Berlin Heidelberg, 2012, pp. 25-39. |
| [2] | R.C. Buck, J. Franklin, U. Berger, et al., Perfluoroalkyl and polyfluoroalkyl substances in the environment:terminology, classification and origins, Integr. Environ. Assess. Manag. 7(2011) 513-541. |
| [3] | J.S. Wang, Y.T. Zhang, W. Zhang, et al., Association of perfluorooctanoic acid with HDL cholesterol and circulating miR-26b and miR-199-3p in workers of a fluorochemical plant and nearby residents, Environ. Sci. Technol. 46(2012) 9274-9281. |
| [4] | M.J. Lopez-Espinosa, T. Fletcher, B. Armstrong, et al., Association of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS) with age of puberty among children living near a chemical plant, Environ. Sci. Technol. 45(2011) 8160-8166. |
| [5] | B.B. Gump, Q. Wu, A.K. Dumas, K. Kannan, Perfluorochemical (PFC) exposure in children:associations with impaired response inhibition, Environ. Sci. Technol. 45(2011) 8151-8159. |
| [6] | U. S. Environmental Protection Agency (US EPA), New Chemical Review of Alternatives for PFOA and Related Chemicals, 2012 http://www.epa.gov/oppt/pfoa/pubs/altnewchems.html. |
| [7] | ENV/JM/RD (2002) 17/FINAL, Hazard assessment of perfluorooctane sulfonate (PFOS) and its salts. http://www.oecd.org/chemicalsafety/risk-assessment/2382880.pdf. |
| [8] | United Nations Environment Programme, Governments Unite to Step-up Reduction on Global DDT Reliance and Add Nine New Chemicals Under International Treaty, UNEP, 2009 http://chm.pops.int/Convention/Press release COP4 Geneva 8 May 2009/tabid/542/language/en-US/Default.aspx. |
| [9] | Y.N. Liu, A.S. Pereira, S. Beesoon, et al., Temporal trends of perfluorooctanesulfonate isomer and enantiomer patterns in archived Swedish and American serum samples, Environ. Int. 75(2015) 215-222. |
| [10] | Z. Zhou, Y.L. Shi, R. Vestergren, et al., Highly elevated serum concentrations of perfluoroalkyl substances in fishery employees from Tangxun Lake, China, Environ. Sci. Technol. 48(2014) 3864-3874. |
| [11] | J.W. Teng, S.Z. Tang, S.Y. Qu, Determination of perfluorooctanesulfonate and perfluorooctanoate in water samples by SPE-HPLC/electrospray ion trap mass spectrometry, Microchem. J. 93(2009) 55-59. |
| [12] | J.M. Keller, A.M. Calafat, K. Kato, et al., Determination of perfluorinated alkyl acid concentrations in human serum and milk standard reference materials, Anal. Bioanal. Chem. 397(2010) 439-451. |
| [13] | A. Navarrete, M.P. Martínez-Alcázar, I. Durá n, et al., Simultaneous online SPE-HPLC-MS/MS analysis of docetaxel, temsirolimus and sirolimus in whole blood and human plasma, J. Chromatogr. B:Analyt. Technol. Biomed. Life Sci. 921/922(2013) 35-42. |
| [14] | F. Guo, Q. Liu, G.B. Qu, et al., Simultaneous determination of five estrogens and four androgens in water samples by online solid-phase extraction coupled with high-performance liquid chromatography-tandem mass spectrometry, J. Chromatogr. A 1281(2013) 9-18. |
| [15] | F. Gosetti, U. Chiuminatto, D. Zampieri, et al., Determination of perfluorochemicals in biological, environmental and food samples by an automated online solid phase extraction ultra high performance liquid chromatography tandem mass spectrometry method, J. Chromatogr. A 1217(2010) 7864-7872. |
| [16] | P. Zhang, Y.L. Shi, Y.Q. Cai, S.F. Mou, Determination of perfluorinated compounds in water samples by high performance liquid chromatography-electrospray tandem mass spectrometry, Chin. J. Anal. Chem. 35(2007) 969-972. |
| [17] | S. Jiang, Y.S. Li, B. Sun, Determination of trace level of perchlorate in Antarctic snow and ice by ion chromatography coupled with tandem mass spectrometry using an automated sample on-line preconcentration method, Chin. Chem. Lett. 24(2013) 311-314. |