 2015, Vol.26
2015, Vol.26 



Amines are a class of organic compounds that are widespread in natural products, pharmaceuticals and biologically important compounds, and they have attracted extensive synthetic efforts [1]. Because of their high nucleophilicity, basicity and susceptibility to oxidation, amines are usually protected in the course of organic transformations to avoid undesired side reactions. The acylation of amines to amides is one of the most frequently used methods for the protection of amines. The acylated amines (amides) are well utilized in asymmetric hydrogenation of enamides [2], kinetic resolutions [3], and C-H activation reactions [4], as a result of the important protecting/directing role of acyl group. Cleavage of the amide bond, however, is difficult, because of the delocalization of the electrons across the amide. The conventional N-deacylation of amides, such as hydrolysis, alcoholysis and transamidation, are usually performed under harsh reaction conditions, such as use of strong acids or bases, and at high temperature [5]. In the last decade, as the C-H bond activation has progressively become a well-established synthetic tool [4], the development of new methodologies for the facile deprotection of secondary amides has attracted special attention from synthetic chemists. For example, using triphenylphosphite chlorine complex [6] and oxalyl chloride [7] as the activating reagent respectively, Prati and Koenig have developed mild alcoholysis methods for the deacylation of secondary amides. Recently, a catalytic combination system of scandium triflate and boronic ester has been applied by Mashima [8] for the deprotection of acetylaniline derivatives under mild and neutral conditions. Ohshima and Morimoto have recently reported the cleavage of unactivated secondary amides bonds by ammonium salt-accelerated transamidation [9] and hydrazinolysis [10]. Using the Schwartz reagent at room temperature, Bhat [11] has developed a mild and efficient method for the chemoselective deprotection of acetamides in the presence of NBoc or N-Cbz moieties.
In connection with our longstanding interest in the synthesis of bioactive alkaloids [12], our group has been in recent years engaged in the development of new C-C bond formation reactions that employ stable amides as substrates [13]. Charette’s [14] and our group [13b, c] have independently reported the transformation of secondary amides into ketones by alkylation with Grignard and organocerium reagents, respectively (Scheme 1). In this transformation, the stable secondary amides, upon activation by triflic anhydride (Tf2O) [15], are converted to reactive nitrilium ions, which react with organometallic reagents to give imines that are hydrolyzed under acidic conditions to give ketones. It is evident that acidic work-up of imines would produce amines as well. The method could thus, on the other hand, serve as an alternative way for the deprotection of secondary amides to produce primary amines. Using Grignard reagents as alkylation reagents, Charette et al. [14] have showed one example of isolation of amine moiety by an acid/base workup after hydrolysis of the corresponding imine. Such transformation, however, was performed in an extremely diluted dichloromethane solution (0.044 mol/L), which is environmentally harmful, thus preventing the scale-up of the reaction. As a result of recent interest in the development of methods for the deprotection of secondary amides, herein we report our own results on the N-deacylation of secondary amides by alkylation with organocerium reagents.

|
Download:
|
| Scheme 1.Reductive alkylation of secondary amides with Grignard and organocerium reagents. | |
Melting points were uncorrected. Infrared spectra were measured using film KBr pellet techniques. 1H NMR and 13C NMR spectra were recorded on Bruker 400 MHz or 500 MHz spectrometer. Chemical shifts are expressed in δ (ppm) units downfield from TMS. High-resolution mass spectra (HRMS) were recorded on a Bruker APEX II FT mass spectrometer. Silica gel (300-400 mesh) was used for flash column chromatography, eluting (unless other wise stated) with ethyl acetate/n-hexane mixture. Ether and THF were distilled over sodium benzophenone ketyl under N2. Dichloromethane was distilled over calcium hydride under N2. Trifluoromethanesulfonic anhydride (Tf2O) was distilled over phosphorous pentoxide and was stored for no more than a week before redistilling. All other commercially available compounds were used as received. Anhydrous cerium chloride was prepared from CeCl3·7H2O according to the reported procedure [16].
General procedure for N-deacylation of secondary amides: Tf2O (185 μL, 1.1 mmol) was added dropwise to a cooled (0 ℃) solution of amide (1.0 mmol) and 2-fluoropyridine (103 μL, 1.2 mmol) in dichloromethane (4 mL). After stirring at 0 ℃ for 30 min, the mixture was cannulated to a freshly prepared organocerium reagent/complex (3.0 mmol) in THF (15 mL) at -78 ℃ and stirred for 2 h. Aqueous HCl solution (3 mol/L, 5 mL) was added to quench the reaction and the mixture was allowed to warm to r.t. and stirred for 2 h. Ammonium hydroxide solution (25%, 5 mL) was then added to the mixture. The organic layer was separated and the aqueous phase was extracted with diethyl ether (3× 10 mL). The combined organic layers were washed with brine (3× 3 mL) and concentrated under reduced pressure to about 1/3 volume. The residual organic phase was then extracted with aqueous HCl solution (3 mol/L, 3× 5 mL). The separated organic phase was washed with brine (5 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure, and the residue was purified by flash column chromatography on silica gel to afford ketone. The aqueous phases were combined, washed with diethyl ether (5 mL), basified with an ammonium hydroxide solution (25%, 5 mL) and back-extracted with diethyl ether (5× 20 mL). The ether layers were combined, washed with brine (5 mL), dried over anhydrous MgSO4, filtered, acidified with a solution of HCl in ethyl acetate (3 mol/L, 5 mL) and concentrated under reduced pressure to afford the desired amine hydrochloride salt.
The characterization data of amine hydrochlorides 2 and ketone 3 were provided in Supporting information.
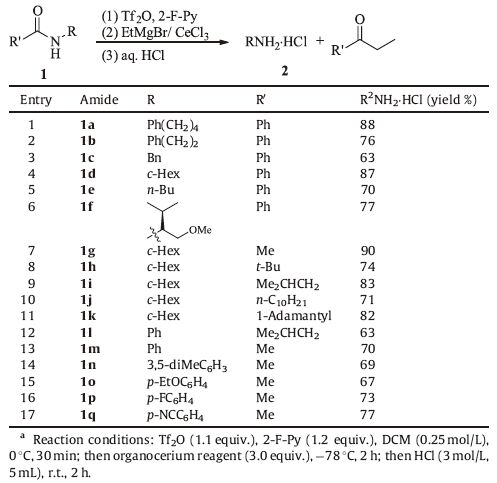
3. Results and discussionOur investigation is based on the direct alkylative deamination of secondaryamides, amethoddeveloped recently fromour laboratory for the synthesis of ketones [13b, c]. The N-deacylation of secondary amide 1a was chosen as the model reaction. Following the general procedure reported for the transformation of secondary amides into ketones [13b, c], a dichloromethane solution of amide 1a and 2-fluoropyridine (1.2 equiv)was treated successively with 1.1 equiv of Tf2O (0 ℃, 30 min) and 3.0 equiv of freshly prepared ethyl cerium regent (-78 ℃, 2 h), and the reaction was quenched with aqueous HCl solution. After work-up through simple acid-base extraction, only 62% of the corresponding primary amine was obtained. In our further trials to improve the yield, we found that there might be somemagnesiumcomplex of amines produced. So after the reaction was quenched with aqueous HCl solution, aqueous ammonia was added to facilitate the liberation of amines from amine-Mg complexes. In view of high volatility of amines, the final products were isolated as amine hydrochloride salts. In this way, the yield of deacylation of 1a was improved to 88%.
With the optimized reaction procedures in hand, we next examined the scope of structurally different benzoylated aliphatic amines (entries 1-6, Table 1). Various acylated primary amines (1a-f) were deprotected to afford the corresponding amine hydrochloride salts 2a-f in good yield upon isolation. It should be mentioned that chiral substrates 1f, which has an α-chiral center, underwent facile N-deacylation smoothly to afford the corresponding amine hydrochloride salt 2f without any epimerization (ee value was determined by HPLC, for details see Supporting information).
|
|
Table 1 Deacylation of secondary amides.a |
{kind=link}
In addition to benzoyl group, other acyl groups such as acetyl, pivaloyl and isovaleroyl groups in 1g-k could be removed to afford amine hydrochloride salts in high yields (entries 7-11).
The reaction conditions could also be applied to the deprotection of a variety of anilides derivatives (entries 12-17). The electronic properties of the functional groups did not have significant impact on the reaction time and yield. Substrates with both electron donating (1n-o) and withdrawing (1p-q) functional moieties underwent N-deacylation rapidly, affording the corresponding amine hydrochloride salts (2n-q) in excellent yields. The deprotection of cyano substituted anilide 1q went chemoselectively to furnish the corresponding p-cyano aniline hydrochloride salt 2q in 77% yield.
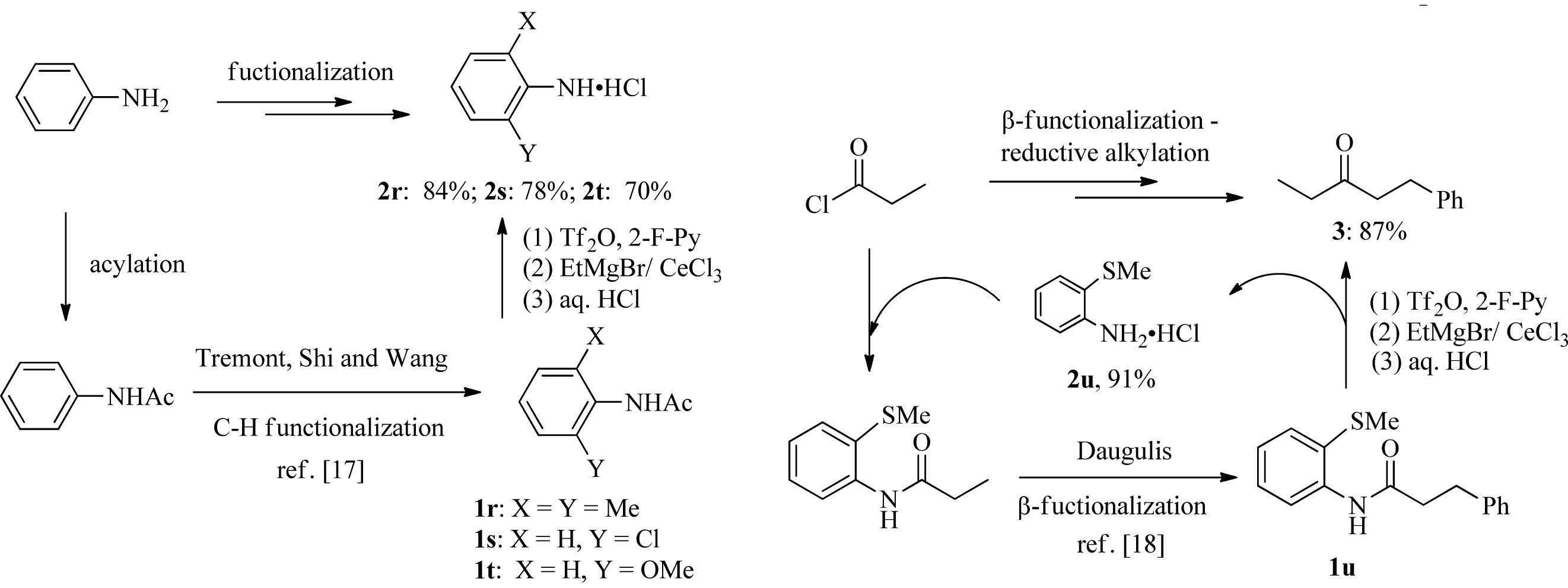
As the secondary amides have been recently used as powerful directing groups in C-H activation/functionalization [4], several acylated amine intermediates obtained from Pd-catalyzed C-H bond activations are examined (Scheme 2). The deprotection of anilines 1r-t, the ortho-functionalization products of aniline reported respectively by Tremont, Shi and Wang [17], proceeded smoothly to give ortho-substituted anilines hydrochloride 2r-t. The deacylation method, in combination with acylation and catalytic C-H bond activations, could thus be used for orthofunctionalization of aniline. The method was also applicable to deacylation of acetanilide 1u, the β-arylation product of sp3 C-H bond reported by Daugulis [18], giving anilines hydrochloride 2u as well as ketone 3 in 91% and 87% yield, respectively. The recovered aniline hydrochloride 2u could be used for the next run of conversion of propionyl halide to ketone 3. It should be mentioned that the steric factor in substrates 1r-u did not have obvious impact on the rate of deprotection.

|
Download:
|
| Scheme 2.Application of the deacylation in combination with functionalization methodologies. | |
{kind=link}
In summary, we have reported a reliable protocol for the N-deacylation of secondary amides by alkylation with organocerium reagents. The deprotection proceeded smoothly at -78 ℃ to produce the corresponding amine hydrochloride salts in good yield. In combination with C-H activation/functionalization, the method is applicable to the functionalization of aniline as well as conversion of carboxylic derivatives to functionalized ketones.
AcknowledgmentsThe authors are grateful for financial support from the National Natural Science Foundation of China (No. 21332007), the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) of Ministry of Education, the Fundamental Research Funds for the Central Universities (No. 20720150044), and the Natural Science Foundation of Fujian Province of China (No. 2014J01062).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2015.05.033.
| [1] | Chiral Amine Synthesis, in:T.C. Nugent (Ed.), Methods, Developments and Applications, Wiley-VCH, Weinheim, 2010. |
| [2] | (a) K. Gopalaiah, H.B. Kagan, Use of nonfunctionalized enamides and enecarbamatesin asymmetric synthesis, Chem. Rev. 111(2011) 4599-4657; (b) J.H. Xie, S.F. Zhu, Q.L. Zhou, Transition metal-catalyzed enantioselective hydrogenation of enamines and imines, Chem. Rev. 111(2011) 1713-1760; (c) T.C. Nugent, M. El-Shazly, Chiral amine synthesis-recent developments and trends for enamide reduction, reductive amination, and imine reduction, Adv. Synth. Catal. 352(2010) 753-819. |
| [3] | (a) E. Busto, V. Gotor-Ferná ndez, V. Gotor, Hydrolases in the stereoselective synthesis of N-heterocyclic amines and amino acid derivatives, Chem. Rev. 111(2011) 3998-4035; (b) C.E. Müller, P.R. Schreiner, Organocatalytic enantioselective acyl transfer onto racemic as well as meso alcohols, amines, and thiols, Angew. Chem. Int. Ed. 50(2011) 6012-6042; (c) H. Pellissier, Catalytic non-enzymatic kinetic resolution, Adv. Synth. Catal. 353(2011) 1613-1666. |
| [4] | (a) P.B. Arockiam, C. Bruneau, P.H. Dixneuf, Ruthenium(II)-catalyzed C-H bond activation and functionalization, Chem. Rev. 112(2012) 5879-5918; (b) T.W. Lyons, M.S. Sanford, Palladium-catalyzed ligand-directed C-H functionalization reactions, Chem. Rev. 110(2010) 1147-1169; (c) L. Ackermann, R. Vicente, A.R. Kapdi, Transition-metal-catalyzed direct arylation of (hetero)arenes by C-H bond cleavage, Angew. Chem. Int. Ed. 48(2009) 9792-9826; (d) X. Chen, K.M. Engle, D.H. Wang, J.Q. Yu, Palladium(II)-catalyzed C-H activation/C-C cross-coupling reactions:versatility and practicality, Angew. Chem. Int. Ed. 48(2009) 5094-5115; (e) O. Daugulis, H.Q. Do, D. Shabashov, Palladium- and copper-catalyzed arylation of carbon-hydrogen bonds, Acc. Chem. Res. 42(2009) 1074-1086. |
| [5] | P.G.M. Wuts, T.W. Greene, Protective Groups in Organic Synthesis, 4th ed., Wiley-Interscience, Hoboken, 2006, pp. 773-789. |
| [6] | A. Spaggiari, L.C. Blaszczak, F. Prati, Low-temperature deacylation of N-monosubstituted amides, Org. Lett. 6(2004) 3885-3888. |
| [7] | S.G. Koenig, C.P. Vandenbossche, H. Zhao, et al., A facile deprotection of secondary acetamides, Org. Lett. 11(2009) 433-436. |
| [8] | Y. Kita, Y. Nishii, A. Onoue, K. Mashima, Combined catalytic system of scandium triflate and boronic ester for amide bond cleavage, Adv. Synth. Catal. 355(2013) 3391-3395. |
| [9] | Y. Shimizu, H. Morimoto, M. Zhang, T. Ohshima, Microwave-assisted deacylation of unactivated amides using ammonium-salt-accelerated transamidation, Angew. Chem. Int. Ed. 51(2012) 8564-8567. |
| [10] | Y. Shimizu, M. Noshita, Y. Mukai, H. Morimoto, T. Ohshima, Cleavage of unactivated amide bonds by ammonium salt-accelerated hydrazinolysis, Chem. Commun. 50(2014) 12623-12625. |
| [11] | P.R. Sultane, T.B. Mete, R.G. Bhat, Chemoselective N-deacetylation under mild conditions, Org. Biomol. Chem. 12(2014) 261-264. |
| [12] | (a) A.E. Wang, P.Q. Huang, Efficient asymmetric syntheses of alkaloids and medicinally relevant molecules based on heterocyclic chiral building blocks, Pure Appl. Chem. 86(2014) 1227-1235; (b) H.Q. Deng, X.Y. Qian, Y.X. Li, et al., A versatile two-step method for the reductive alkylation and formal[4+2] annulation of secondary lactams:step economical syntheses of the ant venom alkaloids cis-2-butyl-5-propylpyrrolidine and (+)-monomorine I, Org. Chem. Front. 1(2014) 258-266; (c) S.P. Luo, L.D. Guo, L.H. Gao, S. Li, P.Q. Huang, Toward the total synthesis of haliclonin A:construction of a tricyclic substructure, Chem. Eur. J. 19(2013) 87-91; (d) X.G. Wang, A.E. Wang, P.Q. Huang, A concise formal stereoselective total synthesis of (-)-swainsonine, Chin. Chem. Lett. 25(2014) 193-196; (e) S.P. Luo, Q.L. Peng, C.P. Xu, A.E. Wang, P.Q. Huang, Bioinspired step-economical, redox-economical and protecting-group-free enantioselective total syntheses of (-)-chaetominine and analogues, Chin. J. Chem. 32(2014) 757-770; (f) P.Q. Huang, S.Y. Huang, L.H. Gao, et al., Enantioselective total synthesis of (+)-methoxystemofoline and (+)-isomethoxystemofoline, Chem. Comm. 51(2015) 4576-4578. |
| [13] | (a) K.J. Xiao, J.M. Luo, K.Y. Ye, Y. Wang, P.Q. Huang, Direct, one-pot sequential reductive alkylation of lactams/amides with Grignard and organolithium reagents through lactam/amide activation, Angew. Chem. Int. Ed. 49(2010) 3037-3040; (b) K.J. Xiao, A.E. Wang, Y.H. Huang, P.Q. Huang, Versatile and direct transformation of secondary amides into ketones by deaminative alkylation with organocerium reagents, Asian J. Org. Chem. 1(2012) 130-132; (c) K.J. Xiao, Y.H. Huang, P.Q. Huang, General direct transformation of secondary amides to ketones via amide activation, Acta Chim. Sinica 70(2012) 1917-1922; (d) Z.Y. Mao, S.Y. Huang, L.H. Gao, A.E. Wang, P.Q. Huang, A novel and versatile method for the enantioselective syntheses of tropane alkaloids, Sci. China Chem. 57(2014) 252-264; (e) P.Q. Huang, W. Ou, K.J. Xiao, A.E. Wang, Tertiary amide-based Knoevenageltype reactions:a direct, general, and chemoselective approach to enaminones, Chem. Comm. 50(2014) 8761-8763; (f) P.Q. Huang, Q.W. Lang, A.E. Wang, J.F. Zheng, Direct reductive coupling of secondary amides:chemoselective formation of vicinal diamines and vicinal amino alcohols, Chem. Comm. 51(2015) 1096-1099; (g) P.Q. Huang, Y. Wang, K.J. Xiao, Y.H. Huang, A general method for the direct transformation of common tertiary amides into ketones and amines by addition of Grignard reagents, Tetrahedron 71(2015) 4248-4254. |
| [14] | W.S. Bechara, G. Pelletier, A.B. Charette, Chemoselective synthesis of ketones and ketimines by addition of organometallic reagents to secondary amides, Nat. Chem. 4(2012) 228-234. |
| [15] | For reviews on the chemistry of triflic acid and its derivatives, see:(a) PJ. Stang, M.R. White, Triflic acid and its derivatives, Aldrichimica Acta 16(1983) 15-22; (b) I.L. Baraznenok, V.G. Nenajdenko, E.S. Balenkova, Chemical transformations induced by triflic anhydride, Tetrahedron 56(2000) 3077-3119; (c) M. Movassaghi, M.D. Hill, O.K. Ahmad, Direct synthesis of pyridine derivatives, J. Am. Chem. Soc. 129(2007) 10096-10097; (d) S.L. Cui, J. Wang, Y.G. Wang, Synthesis of indoles via domino reaction of N-aryl amides and ethyl diazoacetate, J. Am. Chem. Soc. 130(2008) 13526-13527; (e) B. Peng, D. Geerdink, C. Faré s, N. Maulide, Chemoselective intermolecular α-arylation of amides, Angew. Chem. Int. Ed. 53(2014) 5462-5466. |
| [16] | (a) T. Imamoto, Y. Sugiura, N. Takiyama, Organocerium reagents. Nucleophilic addition to easily enolizable ketones, Tetrahedron Lett. 25(1984) 4233-4236; (b) N. Takeda, T. Imamoto, Use of cerium(III) chloride in the reactions of carbonyl compounds with organolithiums or Grignard reagents for the suppression of abnormal reactions:1-butyl-1,2,3,4-tetrahydro-1-naphthol, Org. Synth. 76(1999) 228-238. |
| [17] | (a) S.J. Tremont, H.U. Rahman, Ortho-alkylation of acetanilides using alkyl halides and palladium acetate, J. Am. Chem. Soc. 106(1984) 5759-5760; (b) X.B. Wan, Z.X. Ma, B.J. Li, et al., Highly selective C-H functionalization/halogenation of acetanilide, J. Am. Chem. Soc. 128(2006) 7416-7417; (c) T.S. Jiang, G.W. Wang, Palladium-catalyzed ortho-alkoxylation of anilides via C-H activation, J. Org. Chem. 77(2012) 9504-9509. |
| [18] | D. Shabashov, O. Daugulis, Auxiliary-assisted palladium-catalyzed arylation and alkylation of sp2 and sp3 carbon-hydrogen bonds, J. Am. Chem. Soc. 132(2010) 3965-3972. |