



 , Yu-Zhuo Niu, Ya-Lin Wang, Dan-Feng Huang, Ying-Peng Su, Jin-Xian Wang, Yu-Lai Hu , Ying Fu, Zheng-Yin Du
, Yu-Zhuo Niu, Ya-Lin Wang, Dan-Feng Huang, Ying-Peng Su, Jin-Xian Wang, Yu-Lai Hu , Ying Fu, Zheng-Yin Du
Many methods for preparation of trifluoromethyl tertiary alcohols have been developed. Higashiyama’s group reported the direct catalytic Aldol reaction of trifluoromethyl ketones with ketones to prepare trifluoromethyl tertiary alcohols using diethyl- zinc secondary amine complex as catalysts [6]. Organocatalytic Aldol addition of methyl ketones to aryl trifluoromethyl ketones afforded b-trifluoromethyl-b-hydroxyl ketones in good to excel- lent yield [7]. Although the abovemethods provided easy access to trifluoromethyl tertiary alcohols,it is still necessary to find more environment benign methods.
Solvent free organic reactions have attracted much research interest fromthe point of green chemistry in recent years.Many of solvent free organic reactions were reported to a conversions and yields in short reaction time a temperature [8].
Our research group is engaged in green chemis fluorine chemistry [10] for many years. Herein,we exceedingly fast preparation of trifluoromethyl tertia from Aldol reaction between methyl ketones and trifl ketones in high yields under solvent free conditions.
2. ExperimentalAll reactions were conducted in a 30 mL pear-shaped flask. Reagents were obtained from commercial suppliers and used without further purification unless otherwise noted. Flash column chromatographywas carried out using Qingdao silica gel (230-400 mesh). Analytical thin layer chromatography (TLC) was done using Qingdao silica gel (silica gel GF254). TLC plates were analyzed by an exposure to ultraviolet (UV) light and/or in I2. 1 H NMR, 13 C NMR and 19 F NMR spectra were recorded at 400 MHz and 100 MHz on Varian Mercury 400 plus instrument,respectively. Chemical shifts are reported as d values (ppm) relative to tetramethylsilane (TMS) for 1 H NMR and chloroformfor 13 C NMR. Coupling constants (J) are reported in Hertz (Hz). Melting points were uncorrected. Infrared spectra were recorded on an IR spectrometer (Perkin Elmer BX FT-IR),and absorption frequencies were reported in reciprocal centimeters (cm1 ). The HRMS dataweremeasured onMALDI-TOF type of instrument for the high-resolution mass spectra.
2.1. Synthesis of b-trifluoromethyl-b-hydroxyl ketones 3The mixture of trifluoroacetophenone (34.8 mg,0.2 mmol) and acetophenone (24.0 mg,0.2 mmol)was put into oven-dried,30 mL pear-shaped flask at room temperature,and then lithium hydroxide powder (5.3 mg,0.22 mmol) was added. The mixture was grinded and stirred in the flask at room temperature for 5- 16 min,and then dissolved in water (5 mL) and ethyl acetate (5 mL). The organic phase was separated. Aqueous phase was extracted with ethyl acetate (3×5 mL). The organic layer was combined,dried over anhydrous MgSO4,and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography with petroleum ether and ethyl acetate as eluent to give the pure 3a. Other target products were obtained in the same procedure.
4,4,4-Trifluoro-3-hydroxy-1,3-diphenylbutan-1-one (3a) [6]: White solid,mp: 42-43 8C. 1 H NMR (400 MHz,DMSO-d6): δ 7.95 (d,2H,J = 7.6 Hz),7.61-7.65 (m,3H),7.51 (t,2H,J = 7.2 Hz),7.28- 7.37 (m,3H),6.62 (s,1H),4.27 (d,1H,J = 17.2 Hz),3.83 (d,1H, J = 17.2 Hz). 13 C NMR (100 MHz,DMSO-d6): δ 200.2,143.1,142.1, 138.4,133.7,133.1,132.9,132.8,131.6,130.5 (q,J = 285.0 Hz),80.2 (q,J = 27.2 Hz),46.5. 19 F NMR (376 MHz,CDCl3): δ-77.01.
1-(2-Bromophenyl)-4,4,4-trifluoro-3-hydroxy-3-phenylbutan- 1-one (3b) [7d]: Colorless oil. 1 H NMR (400 MHz,CDCl3): δ 7.58- 7.62 (m,3H),7.26-7.35 (m,5H),7.18-7.19 (m,1H),5.36 (s,1H), 3.96 (d,1H,J = 17.2 Hz),3.74 (d,1H,J = 17.2 Hz). 13 C NMR (100 MHz,CDCl3): δ 203.4,140.5,137.1,133.9,132.6,129.0, 128.8,128.4,127.6,126.4,124.4 (q,J = 283.5 Hz),118.8,76.6 (q, J = 29.1 Hz),45.1. 19 F NMR (376 MHz,CDCl3): δ-80.50.
1-(3-Bromophenyl)-4,4,4-trifluoro-3-hydroxy-3-phenylbutan- 1-one (3c): Colorless oil. 1 H NMR (400 MHz,DMSO-d6): δ 8.07 (s, 1H),7.91 (d,1H,J = 7.2 Hz),7.83 (d,1H,J = 7.6 Hz),7.61 (d,2H, J = 6.8 Hz),7.46 (t,1H,J = 7.6 Hz),7.41-7.23 (m,3H),6.64 (s,1H), 4.28 (d,1H,J = 17.6 Hz),3.80 (d,1H,J = 17.6 Hz). 13 C NMR (100 MHz,DMSO-d6): δ 199.1,144.2,143.0,141.0,136.0,135.8, 133.0,132.9,132.2,131.7,130.5 (q,J = 285.2 Hz),127.2,80.2 (q, J = 27.4 Hz),47.1. 19 F NMR (376 MHz,CDCl3): δ-80.62. HRMS (ESI) Calcd. for C16H12BrF3O2 (M+ Na): 394.9865,Found: 394.9870.
1-(4-Bromophenyl)-4,4,4-trifluoro-3-hydroxy-3-phenylbutan- 1-one (3d) [3]: White solid,mp: 109-110 8C. 1 H NMR (400 MHz, DMSO-d6): δ 7.87 (d,2H,J = 7.6 Hz),7.72 (d,2H,J = 7.2 Hz),7.61 (d, 2H,J = 6.8 Hz),7.30-7.36 (m,3H),6.62 (s,1H),4.24 (d,1H, J = 17.6 Hz),3.79 (d,1H,J = 17.6 Hz). 13 C NMR (100 MHz,DMSO- d6): δ 199.4,143.0,141.2,136.8,135.3,133.0,132.9,132.5,131.6, 130.5 (q,J = 285.0 Hz),80.2 (q,J = 27.3 Hz),46.7. 19 F NMR (376 MHz,DMSO-d6): δ-79.54.
1-(2-Chlorophenyl)-4,4,4-trifluoro-3-hydroxy-3-phenylbutan- 1-one (3e) [7d]: Colorless oil. 1 H NMR (400 MHz,DMSO-d6): d 7.50-7.56 (m,3H),7.42 (s,2H),7.29-7.36 (m,4H),6.82 (s,1H),4.09 (d,1H,J = 16.8 Hz),3.65 (d,1H,J = 16.8 Hz). 13 C NMR (100 MHz, DMSO-d6): δ 197.0,138.7,136.9,132.0,130.1,129.4,129.2,127.9, 127.7,127.0,126.5,125.2 (q,J = 285.4 Hz),75.0 (q,J = 27.4 Hz), 45.9. 19 F NMR (376 MHz,DMSO-d6): δ-79.49.
4,4,4-Trifluoro-3-hydroxy-1-(3-nitrophenyl)-3-phenylbutan- 1-one (3f): Colorless oil. 1 H NMR (400 MHz,DMSO-d6): δ 8.59 (s, 1H),8.43 (d,1H,J = 7.8 Hz),8.33 (d,1H,J = 7.2 Hz),7.78 (t,1H, J = 8.0 Hz),7.61 (d,2H,J = 6.8 Hz),7.23-7.40 (m,3H),6.72 (s,1H), 4.36 (d,1H,J = 17.2 Hz),3.88 (d,1H,J = 17.2 Hz). 13 C NMR (100 MHz,DMSO-d6): δ 198.8,153.1,143.5,142.8,139.5,135.6, 133.1,132.9,132.5,131.7,130.6 (q,J = 284.8 Hz),127.7,80.2 (q, J = 27.7 Hz),47.6. 19 F NMR (376 MHz,DMSO-d6): δ-79.40. HRMS (ESI) Calcd. for C16H12F3NO4 (M + Na): 362.0611,Found: 362.0617.
4,4,4-Trifluoro-3-hydroxy-3-phenyl-1-(p-tolyl)butan-1-one (3g) [6]:White solid,mp: 72-73 8C. 1 H NMR (400 MHz,DMSO-d6): δ 7.83 (d,2H,J = 7.2 Hz),7.59 (d,2H,J = 6.8 Hz),7.28-7.32 (m,5H), 6.56 (s,1H),4.20 (d,1H,J = 17.2 Hz,),3.74 (d,1H,J = 17.2 Hz,),2.35 (s,3H). 13 C NMR (100 MHz,DMSO-d6): δ 194.9,143.8,138.0,134.5, 129.2,128.2,127.8,127.7,126.5,125.4 (q,J = 285.1 Hz),75.2 (q, J = 27.3 Hz),41.1,21.1. 19 F NMR (376 MHz,DMSO-d6): δ-79.53.
4,4,4-Trifluoro-3-hydroxy-1-(4-methoxyphenyl)-3-phenylbu- tan-1-one (3h) [6]: White solid,mp: 90-91 8C. 1 H NMR (400 MHz, DMSO-d6): δ 7.92 (d,2H,J = 7.6 Hz),7.58 (d,2H,J = 6.8 Hz),7.28- 7.34 (m,3H),7.00 (d,2H,J = 7.6 Hz),6.56 (s,1H),4.15 (d,1H, J = 17.2 Hz),3.81 (s,3H),3.70 (d,1H,J = 17.2 Hz). 13 C NMR (100 MHz,DMSO-d6): δ 194.1,163.4,138.1,130.6,129.9,127.8, 127.7,126.5,125.4 (q,J = 285.2 Hz),113.8,75.3 (q,J = 27.3 Hz), 55.6,40.7. 19 F NMR (376 MHz,DMSO-d6): δ-74.74.
4,4,4-Trifluoro-3-hydroxy-1-(2-hydroxyphenyl)-3-phenylbu- tan-1-one (3i) [11]: White solid,mp: 112-113 8C. 1 H NMR (400 MHz,DMSO-d6): δ 11.40 (s,1H),7.84 (d,1H,J = 8.0 Hz), 7.61 (d,2H,J = 6.8 Hz),7.49 (t,1H,J = 7.6 Hz),7.30-7.35 (m,3H), 6.94 (d,2H,J = 7.2 Hz),6.65 (s,1H),4.31 (d,1H,J = 17.6 Hz),3.86 (d, 1H,J = 17.6 Hz). 13 C NMR (100 MHz,DMSO-d6): δ 200.0,160.2, 138.1,136.1,131.0,128.1,128.0,126.7,125.6 (q,J = 285.5 Hz), 122.0,119.4,117.8,75.3 (q,J = 27.0 Hz),43.1. 19 F NMR (376 MHz, DMSO-d6): δ-77.01.
1-([1,10 -Biphenyl]-4-yl)-4,4,4-trifluoro-3-hydroxy-3-phenyl- butan-1-one (3j): White solid,mp: 51-52 8C. 1 HNMR(400MHz, DMSO-d6): δ 8.02 (d,2H,J = 8.0 Hz),7.79 (d,2H,J = 8.0 Hz),7.72 (d,2H,J = 7.2 Hz),7.63 (d,2H,J =7.2Hz),7.49(t,2H,J =7.2Hz), 7.40-7.43 (m,1H),7.29-7.36 (m,3H),6.64 (s,1H),4.27 (d,1H, J = 17.2 Hz),3.83 (d,1H,J = 17.2 Hz). 13 CNMR(100MHz,DMSO- d6): δ 199.9,149.8,143.9,143.1,140.9,134.2,133.9,133.5,132.9, 132.8,132.1,131.9,131.6,130.5 (q,J = 285.3 Hz),80.3 (q, J = 27.3 Hz),46.5. 19 F NMR (376 MHz,DMSO-d6): δ-79.46. HRMS (ESI) Calcd. for C22H17F3O2 (M + Na): 393.1073,Found: 393.1078.
4,4,4-Trifluoro-3-hydroxy-1-(5-methylfuran-2-yl)-3-phenyl- butan-1-one (3k): Colorless oil. 1 H NMR (400 MHz,DMSO-d6): d 7.61 (d,2H,J = 6.4 Hz),7.51 (s,1H),7.34 (d,3H,J = 7.6 Hz),6.67 (s, 1H),6.36 (s,1H),3.88 (d,1H,J = 16.0 Hz),3.51 (d,1H,J = 16.0 Hz), 2.33 (s,3H). 13 C NMR (100 MHz,DMSO-d6): δ 182.6,158.2,150.9, 137.5,128.0,127.7,126.5,125.2 (q,J = 284.8 Hz),121.5,109.4,75.2 (q,J = 27.5 Hz),40.7,13.5. 19 F NMR (376 MHz,DMSO-d6): δ-79.34. HRMS (ESI) Calcd. for C15H13F3O3 (M + Na): 321.0709,Found: 321.0714.
4,4,4-Trifluoro-3-hydroxy-3-phenyl-1-(pyridin-3-yl)butan-1- one (3l): Colorless oil. 1 H NMR (400 MHz,DMSO-d6): δ 9.05 (s,1H), 8.75 (d,1H,J = 4.0 Hz),8.22 (d,1H,J = 7.2 Hz),7.59 (d,2H, J = 6.8 Hz),7.48-7.52 (m,1H),7.28-7.32 (m,3H),6.67 (s,1H),4.25 (d,1H,J = 17.2 Hz),3.83 (d,1H,J = 17.2 Hz). 13 C NMR (100 MHz, DMSO-d6): δ 199.7,158.5,154.4,142.9,140.7,137.5,133.0,132.9, 131.6,130.5 (q,J = 285.2 Hz),128.8,80.2 (q,J = 27.3 Hz),47.3. 19 F NMR (376 MHz,DMSO-d6): δ-79.36. HRMS (ESI) Calcd. for C15H12F3NO2 (M + H): 296.0893,Found: 296.0898.
1-(2,5-Dimethylthiophen-3-yl)-4,4,4-trifluoro-3-hydroxy-3- phenylbutan-1-one (3m): Colorless oil. 1 H NMR (400 MHz,DMSO- d6): δ 7.59 (d,2H,J = 7.2 Hz),7.32-7.37 (m,4H),6.54 (s,1H),4.01 (d, 1H,J = 17.2 Hz),3.57 (d,1H,J = 17.2 Hz),2.43 (s,3H),2.39 (s,3H). 13 C NMR (100 MHz,DMSO-d6): δ 196.5,151.5,143.1,141.0,139.9, 132.9,132.8,131.9,131.6,130.4 (q,J = 285.5 Hz),80.3 (q, J = 26.7 Hz),49.0,20.6,19.7. 19 F NMR (376 MHz,DMSO-d6): δ-74.87. HRMS (ESI) Calcd. for C16H15F3O2S (M + H): 329.0818, Found: 329.0824.
4,4,4-Trifluoro-3-hydroxy-3-(4-nitrophenyl)-1-phenylbutan- 1-one (3n): Colorless oil. 1 H NMR (400 MHz,DMSO-d6): δ 8.20 (d, 2H,J = 8.0 Hz),7.90-7.92 (m,4H),7.60-7.62 (m,1H),7.48-7.51 (m, 2H),7.02 (s,1H),4.41 (d,1H,J = 18.0 Hz),3.94 (d,1H,J = 18.0 Hz). 13 C NMR (100 MHz,DMSO-d6): δ 199.1,152.2,151.0,141.7,138.5, 133.7,133.1,130.2 (q,J = 285.4 Hz),127.9,79.9 (q,J = 27.2 Hz), 46.8. 19 F NMR (376 MHz,DMSO-d6): δ-79.40. HRMS (ESI) Calcd. for C16H12F3NO4 (M + H): 340.0791,Found: 340.0796.
3-(3-Bromophenyl)-4,4,4-trifluoro-3-hydroxy-1-phenylbutan- 1-one (3o) [7d]: Colorless oil. 1 H NMR (400 MHz,DMSO-d6): δ 7.92 (d,2H,J = 7.6 Hz),7.82 (s,1H),7.61 (d,2H,J = 5.6 Hz),7.49-7.51 (m, 3H),7.27-7.31 (m,1H),6.79 (s,1H),4.31 (d,1H,J = 17.6 Hz),3.83 (d,1H,J = 17.6 Hz). 13 C NMR (100 MHz,DMSO-d6): δ 194.4,140.8, 136.8,133.3,130.6,129.8,129.3,128.5,128.0,125.5,125.1 (q,J = 285.2 Hz),121.3,74.5 (q,J = 27.3 Hz),41.3. 19 F NMR (376 MHz,DMSO-d6): δ-79.70.
3-(4-Chlorophenyl)-4,4,4-trifluoro-3-hydroxy-1-phenylbutan- 1-one (3p) [6]: White solid,mp: 71-73 8C. 1 H NMR (400 MHz, DMSO-d6): δ 7.94 (d,2H,J = 7.2 Hz),7.64-7.66 (m,3H),7.50-7.53 (m,2H),7.42 (d,2H,J = 7.2 Hz),6.78 (s,1H),4.30 (d,1H, J = 17.6 Hz),3.86 (d,1H,J = 17.6 Hz). 13 C NMR (100 MHz,DMSO- d6): δ 194.5,137.1,136.8,133.3,132.6,128.5,128.4,127.9,127.6, 125.2 (q,J = 285.3 Hz),74.6 (q,J = 27.8 Hz),41.2. 19 F NMR (376 MHz,DMSO-d6): δ-79.82.
4,4,4-Trifluoro-3-hydroxy-3-(4-methoxyphenyl)-1-phenylbu- tan-1-one (3q) [6]: White solid,mp: 71-72 8C. 1 H NMR (400 MHz, DMSO-d6): δ 7.93 (d,2H,J = 7.2 Hz,),7.60-7.61 (m,1H),7.49-7.51 (m,4H),6.88 (d,2H,J = 8.4 Hz),6.51 (s,1H),4.18 (d,1H, J = 17.2 Hz),3.75 (d,1H,J = 17.2 Hz),3.72 (s,3H). 13 C NMR (100 MHz,DMSO-d6): δ 195.4,158.8,137.0,133.3,129.7,128.6, 128.0,127.8,125.4 (q,J = 284.8 Hz),113.1,74.9 (q,J = 27.5 Hz), 55.0,41.2. 19 F NMR (376 MHz,DMSO-d6): δ-79.84.
4,4,4-Trifluoro-3-hydroxy-1-phenyl-3-(p-tolyl)butan-1-one (3r) [6]: Colorless oil. 1 H NMR (400 MHz,DMSO-d6): δ 7.94 (d,2H, J = 6.8 Hz),7.62-7.63 (m,1H),7.50 (s,4H),7.14 (d,2H,J = 7.2 Hz), 6.54 (s,1H),4.23 (d,1H,J = 17.2 Hz),3.77 (d,1H,J = 17.2 Hz),2.28 (s,3H). 13 C NMR (100 MHz,DMSO-d6): δ 195.2,137.0,134.9,133.3, 128.6,128.3,128.0,126.4,125.4 (q,J = 285.0 Hz),75.0 (q, J = 27.0 Hz),41.2,20.5. 19 F NMR (376 MHz,DMSO-d6): δ-79.73.
4,4,4-Trifluoro-3-hydroxy-3-(naphthalen-1-yl)-1-phenylbu- tan-1-one (3s) [7d]:White solid,mp: 80-81 8C. 1 H NMR (400 MHz, DMSO-d6): δ 9.16 (s,1H),8.07 (d,2H,J = 7.6 Hz),7.97-8.02 (m,2H), 7.74-7.75 (m,2H),7.66-7.52 (m,4H),7.46-7.48 (m,1H),6.99 (s, 1H),4.88 (d,1H,J = 18.4 Hz),4.00 (d,1H,J = 18.4 Hz). 13 C NMR (100 MHz,DMSO-d6): δ 194.9,136.8,134.3,133.4,133.1,131.9, 129.5,128.6,128.5,127.9,127.6,126.7,126.0 (q,J = 286.2 Hz), 125.3,125.1,124.4,78.4 (q,J = 28.9 Hz),43.2. 19 F NMR (376 MHz, DMSO-d6): δ-78.07.
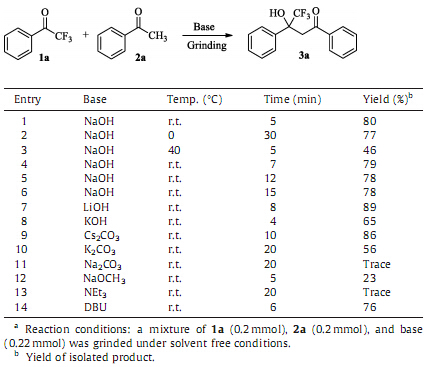
3. Results and discussionAlthough many methods for preparation of trifluoromethyl organic compounds have been developed by using trifluoromethy- lation reagents,such as Togni reagent,Umemoto reagent,and Ruppert-Prakash reagent,trifluoromethyl group-containing build- ingblocks still play important roles inpreparationof trifluoromethyl compounds because of their easy commercial availability,low cost and stability [12]. The Aldol reaction using trifluoromethyl ketones were apparently a convergent method for preparation of trifluor- omethyl tertiary alcohols. Thus,we firstly tried the Aldol reaction between trifluoroacetophenone 1a and acetophenone 2a in the presence of NaOH without any catalyst at room temperature under solvent free conditions to get b-trifluoromethyl-b-hydroxyl ketones. The reaction proceeded smoothly to generate Aldol product 4,4,4-trifluoro-3-hydroxy-1,3-diphenylbutan-1-one 3a with the yield of 80%. However,if the ethanol was used as solvent,there was no reaction occurred. It was found that the reaction was exothermic under grinding condition with liquification of the reaction mixture,and then solidification of the product 3a [13].
In order to improve the yields of the products,the reaction temperature was firstly examined. It was found that the tempera- ture had great influence on the reaction. When the reaction was carriedout at roomtemperature,the product 3acouldbe obtained in 80% yield (Table 1,entry 1). Lowering reaction temperature would prolong the reaction time (Table 1,entry 2).However,the yield of 3a was only 46% with the appearance of ethanol-insoluble solid when the reaction temperature was increased to 40 8C (Table 1,entry 3). The effects of reaction time were examined next. The results demonstrated that 5 minwas enough for completion of the reaction (Table 1,entries 4-6). When the reaction time was prolonged,the yield of 3awill not change toomuch. The choice of base was crucial to obtain good yields (Table 1,entries 7-14). A series of bases were tested,it was found that LiOH was the best base (Table 1,entry 7). Thus,the best result was achieved in the presence of LiOH at room temperature under solvent free conditions (Table 1,entry 7).
| Table 1 Optimization of reaction conditions for the synthesis of 3a.a |
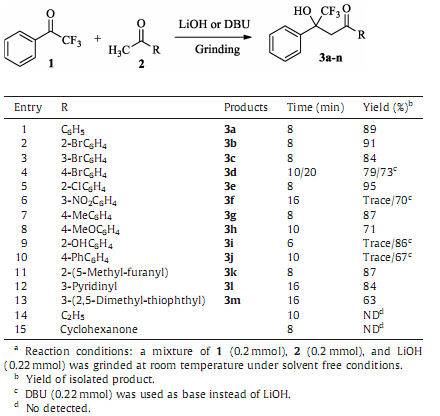
In order to demonstrate the efficiency and the applicability of the method,the reactions were performed with various methyl ketones and trifluoromethyl ketones under the optimized condi- tions. The results are summarized in Tables 2 and 3.
It showed that aromatic methyl ketones and trifluoromethyl ketones reacted smoothly affording the corresponding b-trifluor- omethyl-b-hydroxyl ketones in good to excellent yields (63%-95%). Aliphaticmethyl ketones failed to give the desired products (Table 2, entries 14 and 15). This is due to the cross condensations between aliphatic methyl ketones to make the products very complex. The reactions of heterocyclic ketones with trifluoroacetophenone proceeded smoothly to furnish the products in good yields (Table 2,entries 11-13). The other trifluoromethyl ketones could also be converted to the corresponding products (Table 3). No obvious electronic effects of the ketones were observed.
| Table 2 Solvent free synthesis of b-trifluoromethyl-b-hydroxyl ketones from various methyl ketones.a. |
| Table 3 Solvent free synthesis of b-trifluoromethyl-b-hydroxyl ketones from various trifluoromethyl ketones.a. |
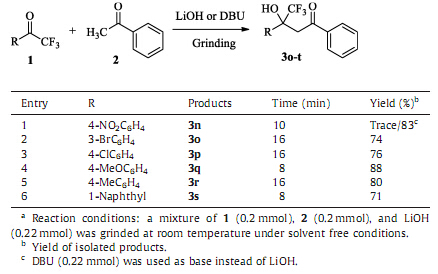
In the course of the experiment,we found thatwhen bothmethyl ketones and trifluoromethyl ketoneswere liquid,the solid base LiOH was the best suitable base to obtain the corresponding products 3 in excellent yields. For example,1-(4-bromophenyl)-ethanone reacted with trifluoroacetophenone smoothly to afford the corresponding compound 3d with the yield of 79% when LiOH was used as base. Liquid organic base such asDBU could also been used in the reaction to obtain reasonable yield of 3d (73%,Table 2,entry 4). If methyl ketones or trifluoromethyl ketones were solid,liquid base such as DBUmust beused inorder to obtain good yields (Table 2,entries 6,9, and 10; Table 3,entry 1).
4. ConclusionIn conclusion,b-trifluoromethyl-b-hydroxyl ketones were successfully synthesized from cross Aldol reaction of methyl ketones and trifluoromethyl ketones under solvent free conditions. The reaction could be achieved smoothly with various aromatic methyl ketones,even with some heterocyclic and aliphatic ketone. The features of this procedure are mild conditions,high yields, operational simplicity,and the environmental friendliness.
Acknowledgments
We are thankful for the financial support from the National Natural Science Foundation of China (No. 21262031),Bioactive Product Engineering Research Center for Gansu Distinctive Plants, and State Key Laboratory of Applied Organic Chemistry,Lanzhou University.
| [1] | (a) R. Filler, Y. Kobayashi, L.M. Yagupolskii, Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications, Elsevier, Amsterdam, 1993; (b) R.E. Banks, B.E. Smart, C.J. Tatlow, Organofluorine Chemistry: Principles and Comercial Applications, Springer, New York, 1994, pp. 237-262; (c) T. Hiyama, Organofluorin Compounds: Chemistry and Application, Springer, Berlin, 2000, pp. 183-234; (d) W.P. Gu, J.H. Lin, J.C. Xiao, Direct N-gem-difluorocyclopropylation of nitroheterocycles by utilizing gem-difluorocyclopropyl tosylate, Chin. Chem. Lett. 25 (2014) 24-28. |
| [2] | (a) K. Mü ller, C. Faeh, F. Diederich, Fluorine in pharmaceuticals: looking beyond intuition, Science 317 (2007) 1881-1886; (b) S. Purser, P.R. Moore, S. Swallow, V. Gouverneur, Fluorine in medicinal chemistry, Chem. Soc. Rev. 37 (2008) 320-330; (c) W.K. Hagmann, The many roles for fluorine in medicinal chemistry, J. Med. Chem. 51 (2008) 4359-4369. |
| [3] | H. Kawai, S. Okusu, E. Tokunaga, N. Shibata, Enantioselective synthesis of 5-trifluoromethyl-2-isoxazolines and their N-oxides by [hydroxy(tosyloxy)iodo] benzene-mediated oxidative N-O coupling, Eur. J. Org. Chem. 29 (2013) 6506-6509. |
| [4] | P.V. Ramachandran, Asymmetric Fluoroorganic Chemistry: synthesis, Application and Future Directions, ACS, Washington, 1999, pp. 255-269. |
| [5] | (a) J. Ren, J. Milton, K.L. Weaver, S.A. Short, D.I. Stuart, D.K. Stammers, Structural basis for the resilience of efavirenz (DMP-266) to drug resistance mutations in HIV-1 reverse transcriptase, Structure 8 (2000) 1089-1094; (b) O.S. Pedersen, E.B. Pedersen, The flourishing syntheses of non-nucleoside reverse transcriptase inhibitors, Synthesis 4 (2000) 479-495. |
| [6] | S. Sasaki, K. Kikuchi, T. Yamuchi, K. Higashiyama, Direct Aldol reaction of trifluoromethyl ketones with ketone catalyzed by Et2Zn and secondary amines, Synlett 10 (2011) 1431-1434. |
| [7] | (a) L.H. Qiu, Z.X. Shen, C.Q. Shi, Y.H. Liu, Y.W. Zhang, Proline catalyzed asymmetric Aldol reaction between methyl ketones and 1-aryl-2 2,2-trifluoroethanones, Chin. J. Chem. 23 (2005) 584-588; (b) N. Duangdee, W. Harnying, G. Rulli, et al., Highly enantioselective organocatalytic trifluoromethyl carbinol synthesis -a caveat on reaction times and product isolation, J. Am. Chem. Soc. 134 (2012) 11196-11205; (c) N. Hara, R. Tamura, Y. Funahashi, S. Nakamura, N-(heteroarenesulfonyl) prolinamides-catalyzed Aldol reaction between acetone and aryl trihalomethyl ketones, Org. Lett. 13 (2011) 1662-1665; (d) Y. Zheng, H.Y. Xiong, J. Nie, M.Q. Hua, J.A. Ma, Biomimetic catalytic enantioselective decarboxylative Aldol reaction of β-keto acids with trifluoromethyl ketones, Chem. Commun. 48 (2012) 4308-4310. |
| [8] | (a) M.A.P. Martins, C.P. Frizzo, D.N. Moreira, L. Buriol, P. Machado, Solvent-free heterocyclic synthesis, Chem. Rev. 109 (2009) 4140-4182; (b) G. Choudhary, R. Krishna Peddinti, An expeditious, highly efficient, catalystfree and solvent-free synthesis of nitroamines and nitrosulfides by Michael addition, Green Chem. 13 (2011) 276-282; (c) A. Kumar, S. Sharma, A grinding-induced catalyst-and solvent-free synthesis of highly functionalized 1,4-dihydropyridines via a domino multicomponent reaction, Green Chem. 13 (2011) 2017-2020; (d) D. Wang, J. Li, N. Li, T. Gao, S. Hou, B. Chen, An efficient approach to homocoupling of terminal alkynes: solvent-free synthesis of 1,3-diynes using catalytic Cu (II) and base, Green Chem. 12 (2010) 45-48; (e) B.R. Vaddula, R.S. Varma, J. Leazer, Mixing with microwaves: solvent-free and catalyst-free synthesis of pyrazoles and diazepines, Tetrahedron Lett. 54 (2013) 1538-1541; (f) J. Yang, N. Li, S. Li, W. Wang, L. Li, A. Wang, X. Wang, Synthesis of diesel and jet fuel range alkanes with furfural and ketones from lignocellulose under solvent free conditions, Green Chem. 16 (2014) 4879-4884; (g) S. Yan, Y. Chen, L. Liu, N. He, J. Lin, Three-component solvent-free synthesis of highly substituted bicyclic pyridines containing a ring-junction nitrogen, Green Chem. 12 (2010) 2043-2052; (h) Y.H. Ma, G. Wu, N. Jiang, et al., Microwave-assisted, facile, rapid and solventfree one pot two-component synthesis of some special acylals, Chin. Chem. Lett. 26 (2015) 81-84. |
| [9] | (a) H.W. Zhan, J.X. Wang, X.T. Wang, Solvent-and catalyst-free synthesis of dihydropyrimidinthiones in one-pot under focused microwave irradiation conditions, Chin. Chem. Lett. 19 (2008) 1183-1185; (b) X.Q. Men, T.J. Meng, J.X. Wang, L. Xin, Pd(II) catalyzed addition reaction of benzylzinc bromides or allylzinc iodines with aromatic aldehydes, Chin. J. Org. Chem. 27 (2007) 272-275; (c) J.X. Wang, N. An, Solvent-free synthesis of 1 4-bis(3-aryl-3-oxo-3-propenyl)-benzenes under grind condition, J. Northw. Norm. Univ. (Nat. Sci.) 47 (2011) 59-62. |
| [10] | J. Xu, Y. Hu, D. Huang, et al., Thiourea-catalyzed enantioselective fluorination ofbketo esters, Adv. Synth. Catal. 354 (2012) 515-526. |
| [11] | V.Y. Sosnovskikh, I.S. Ovsyannikov, I.A. Aleksandrova, Ketone-ketone condensation with the participation of polyhaloalkyl phenyl ketones, Zh. Org. Khim. 28 (1992) 518-526. |
| [12] | J. Nie, H.C. Guo, D. Cahard, J.A. Ma, Asymmetric construction of stereogenic carbon centers featuring a trifluoromethyl group from prochiral trifluoromethylated substrates, Chem. Rev. 111 (2011) 455-529. |
| [13] | (a) G. Rothenberg, A.P. Downie, C.L. Raston, J.L. Scott, Understanding solid/solid organic reactions, J. Am. Chem. Soc. 123 (2001) 8701-8708; (b) T. Friščić , W. Jones, Recent advances in understanding the mechanism of cocrystal formation via grinding, Cryst. Growth Des. 9 (2009) 1621-1637; (c) P.R. Patil, K.P.R. Kartha, Application of ball milling technology to carbohydrate reactions: I. Regioselective primary hydroxyl protection of hexosides and nucleoside by planetary ball milling, J. Carbohydr. Chem. 27 (2008) 279-293. |