




b Young Researchers and Elite Club, Central Tehran Branch, Islamic Azad University, Tehran, Iran
Graphene and similar ‘‘two-dimensional’’ materials have attracted increasing technological and scientific interests because of their special mechanical and electronic features [1, 2, 3]. Molecu- lar interaction at their surfaces is a subfield of considerable interest due to potential applications such as electronic devices and chemical sensors [4, 5]. Their high surface area to volume ratio is ideal for gas sensors. The reactivity of graphene is often adjusted by doping with other elements or topological defects [6]. Hexagonal BN,a layered material,so-called ‘‘white graphene’’,has been isolated from bulk BN and could be useful as a complementary two-dimensional dielectric substrate for graphene electronics [7]. In contrast to the half-metal behavior of graphene,BN sheets possess polar B-N bonds and a wide band gap [8]. Zhang et al. [9] have investigated adsorption mechanisms of carbon monoxide on BN sheets with various modifications,including Al doping,mono- vacancies,and Stone-Wales defects via density functional theory (DFT). It was found that the modified sheet is more sensitive than the pristine sheet for detecting CO molecules. In this work,by means of DFT calculations,we investigate the adsorption of N2O molecules on pristine and Al-doped BN sheets to verify whether or not the sheets can be used as gas sensors in environmental monitoring. Besides the interest in N2O as a greenhouse gas,there is also a need tomonitor N2O gas inmedical,dental,and veterinary environments,where N2O is routinely used as an anesthetic. Long- term exposure to N2O gas has been linked to DNA damage, suppression of immune defenses,and reduced fertility [10].
2. Theory and computational methodWe selected a BN sheet,consisting of 33 B and 33 N atoms (Fig. 1) in which the end atoms were saturated with hydrogen atoms to reduce the boundary effects.We also replaced a B atomof the sheet with an Al atom and studied the N2O adsorption on this surface according to the full geometries shown in Figs. 2 and 3. Geometry optimizations were performed using the B3LYP func- tional augmented with an empirical dispersion term (B3LYP-D), with the 6-31G (d) basis set as implemented in the GAMESS suite of programs [11]. The optimization was performed using default convergence criteria in GAMESS with no restrictions.

|
Download:
|
Fig. 1.Optimized structure of the pristine BN nanosheet and N2O/BN sheet complex.
Distances are in
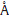 ..
..
|
|
Energy calculations,natural bond orbital analysis (NBO),and density of states (DOS) analysis were performed at the same level of theory. GaussSum program[12] was used to obtain DOS results. The B3LYP density functional has been previously shown to reproduce experimental properties and has been commonly used in nanostructure studies [13, 14, 15, 16]. It has been also demonstrated that the B3LYP provides an efficient and robust basis for calculations of III-V semiconductors by Tomic et al. [17],capable of reliably predicting both the ground state energies and the electronic structure. We used the definition of N2O adsorption energy Ead as follows:
where E(N2O/sheet) corresponds to the energy of the sheet in which the N2O has been adsorbed on the surface,E(sheet) is the energy of the isolated sheet,E(N2O) is the energy of a single N2O molecule,and EBSSE is the energy of the basis set superposition error. The BSSE is calculated based on the Boys and Bernardi counterpoise method [18]. HOMO-LUMO energy gap is defined as where ELUMO and EHOMO are the energies of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO),respectively. 3. Results and discussion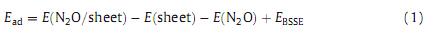

The optimized geometry of the pristine BN sheet is shown in
Fig. 1,in which B-N bond length is in the range 1.460-1.471
,in
good accordance with previous results [19]. The NBO charge
analysis indicates that some electron charges (about 0.49-0.53 e)
are transferred from the B atom to its adjacent N atom within the
sheet,indicating the partially ionic character of the B-N bonds. It
has been previously shown that N2O molecules are weakly
adsorbed on the pristine and Stone-Wales defected BN
nanosheets,indicating that the electronic properties of the sheets
is not sensitive to N2O [20]. Herein,several possible initial
adsorption mechanisms were provided including single (nitrogen
or oxygen),double (N-N or N-O),and triple N-N-O bonded atoms
on the B and N atoms of the sheet. After relaxation,one stable
structure was obtained as shown in Fig. 1,in which the closet
distance between N2O and the sheet is about 3.734
. Our
calculations show that the Ead of N2O is about -5.12 kJ/mol,
indicating a weak adsorption. The calculated energy gaps of the
pristine and N2O adsorbed sheet are about 5.93 and 5.87 eV,
respectively. A chemical sensor works based on the electrical
conductivity change upon the adsorption of an adsorbate. The
electrical conductivity can be related to the energy gap according
to the following equation [21]:

In order to improve the electrical sensitivity of the nanosheet,
we decided to replace a B atomof the sheet with a larger andmore
polarizable Al atom. Experimentally,Hjiri et al. [22] have shown
that Al-doping makes the ZnO nanostructures sensitive to CO
molecules. Inserting an Al atom dramatically changes the
geometric structure of the sheet (Fig. 2). The Al atom is projected
out of the sheet surface in order to reduce stress due to its larger
size compared to a B atom. The formed Al-N bond length is about
1.743
,which is much longer than the corresponding B-N bond.
Subsequently,we explored N2O adsorption on the Al-doped BN
sheet (ABN) by locating themolecule above the Al atomfromits N-
or O-head. After relax optimization,two stable complexes were
predicted (Fig. 3).

|
Download:
|
|
Fig. 2.Optimized structure of Al-doped BN nanosheet and its density of states plot. Distance is in
.
|
|

|
Download:
|
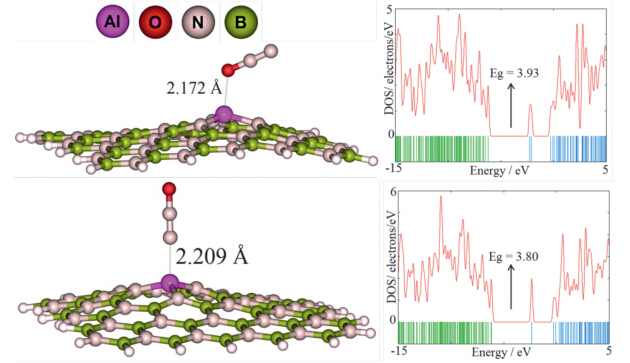
| Fig. 3Optimized structures of N2 O/Al-doped BN nanosheets and their density of states plots. | |
The most stable complex (O) is that in which the N2O molecule
is attached from its oxygen atom to the dopant Al atom. The
predicted Ead for this adsorption process is about -40.86 kJ/mol,indicating that the interaction of N2O molecule with the ABN is
much stronger than thatwith the pristine sheet. The newly formed
Al-O bond length is about 2.172
. The bond lengths of N-Nand N-O of N2O in the complex are about 1.142 and 1.210
,compared to
1.151
and 1.221
for the freemolecule,respectively. Shortening
of these bonds may be because of an NBO charge transfer of about
0.213 e from the molecule to the nanosheet. In the second most
stable configuration (N,Fig. 3),the N2O is attached fromits N atom
to the Al atom with an interaction distance of about 2.209
. The
Ead is about -33.96 kJ/mol,and the charge transfer from the N2O
molecule to the nanosheet is about 0.206 e.
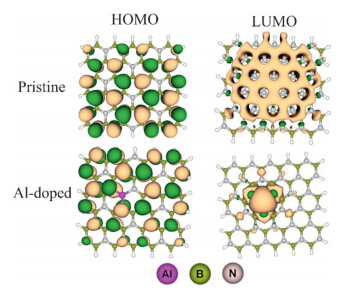
The stronger interaction of the N2O molecule fromits O atomin comparison to the N atom can be rationalized based on the higher NBO negative charge (-0.323 e) which is located on the O atom in comparison to the N head (-0.057 e). It emphasizes that the O- head is a stronger Lewis base in comparison to the N-head. The NBO analysis shows that the hybridization of Al atom is about sp2.94 (nearly sp3 ),in comparison to sp2.01 of the B atom. Thus,the Al atomis projected out and is favorable for nucleophilic attacks. In the other words,unlike boron atoms,the Al atom can have a coordination number of 4 because of its approximately sp3 hybridization. This may be a reason that the interaction of N2O molecule with the ABN is much stronger than the pristine sheet.Also,to determine the nature of this stronger interaction,we analyzed the frontier molecular orbitals. Fig. 4 shows that in the pristine sheet the HOMO and LUMO are localized on the N and B atoms,respectively. After the Al doping,the LUMO is mainly located on the Al atom,but the HOMO is still on the N atoms. Therefore,it is expected that the interaction of the HOMO of the N2O molecule with the LUMO of ABN is stronger than thatwith the LUMO of the pristine sheet.

|
Download:
|
| Fig. 4.The HOMO and LUMO profiles of the pristine and Al-doped BN nanosheets. | |
Calculated DOS of ABN is shown in Fig. 3,indicating that its energy gap value is reduced to 5.50 eV compared to the pristine sheet (5.93 eV). The DOS plot shows that the ABN is still a large-gap semiconductor. DOS plot of the O and N complexes shows a considerable change. New states have appeared at -2.26 and -2.35 eV energy levels in the DOS of O and N complexes in comparison to the bare ABN. This indicates that the electronic properties of the ABN are sensitive to the N2O adsorption. It is revealed from DOS plot of this configuration that its conduction level shifts to lower energies significantly,but the valence level remains constant compared to the bare ABN. The energy gap value of the ABN decreased from 5.50 eV to 3.93 eV and 3.80 eV in the O and N complexes,respectively. These changes would result in an electrical conductivity change of the defected sheet according to Eq. (3). The ABN becomes a p-type semiconductor due to the appearance of an acceptor state within the energy gap after N2O adsorption. Compared with the pristine BN sheet,the ABN would have excellent N2O detection ability. So,we believe that Al doping process may be a good strategy for improving the sensitivity and reactivity of the sheet to N2O,which cannot be trapped and detected by the pristine BN graphene (Table 1).
| Table 1 Calculated energy (Ead,kJ/mol),HOMO energies (EHOMO),LUMO energies (ELUMO),and HOMO-LUMO energy gap (Eg) of the systems in eV. |

We think that the adsorption of N2O on the surface of ABN may be reversible because of themoderate Ead. Very strong interactions are not favorable in gas detection because these imply that desorption could be difficult and the device may suffer from long recovery times. If the Ead significantly becomes more negative, much longer recovery time is expected based on the conventional transition state theory [21]:
where t is recovery time,T is the temperature,k is Boltzmann’s constant,and n0 is the attempt frequency. According to this equation,more negative Ead values will prolong the recovery time in an exponential manner. However,the calculated Ead of N2O in the ABN systemis not so large as hinder the recovery of the sensor, and the recovery time may be short. 4. Conclusion
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The geometric structures and electronic properties of the pristine and Al-doped BN sheets in the presence and absence of an adsorbed N2O moleculewere explored using DFT. Itwas found that the N2O molecule interacts with the pristine BN sheet via van der Waals forces,but it presents much higher reactivity toward the ABN. Based on DOS analysis,it was suggested that ABN may be used in a gas sensor to detect and monitor N2O. The detection mechanism is by an adsorption process which induces a change in conductance of the ABN,providing an electrical signal.
| [1] | K.S. Novoselov, A.K. Geim, S.V. Morozov, et al., Electric field effect in atomically thin carbon films, Science 306 (2004) 666-669. |
| [2] | S.P. Zhang, B. Liu, C.Y. Li, et al., Enhanced dispersibility and thermal stability of β-cyclodextrin functionalized graphene, Chin. Chem. Lett. 25 (2014) 355-358. |
| [3] | Z.L. Cheng, X.X. Qin, Study on friction performance of graphene-based semi-solid grease, Chin. Chem. Lett. 25 (2014) 1305-1307. |
| [4] | J. Beheshtian, H. Soleymanabadi, A.A. Peyghan, Z. Bagheri, A DFT study on the functionalization of a BN nanosheet with PC single bond X, (PC = phenyl carbamate, X = OCH3, CH3, NH2, NO2 and CN), Appl. Surf. Sci. 268 (2013) 436-441. |
| [5] | J. Beheshtian, A.A. Peyghan, Z. Bagheri, Functionalization of BN nanosheet with N2H4 may be feasible in the presence of Stone-Wales defect, Struct. Chem. 24 (2013) 1565-1570. |
| [6] | Y.N. Xu, W.Y. Ching, Calculation of ground-state and optical properties of boron nitrides in the hexagonal, cubic, and wurtzite structures, Phys. Rev. B 44 (1991) 7787. |
| [7] | L. Song, L.J. Ci, H. Lu, et al., Large scale growth and characterization of atomic hexagonal boron nitride layers, Nano Lett. 10 (2010) 3209-3215. |
| [8] | J.Y. Dai, P. Giannozzi, J.M. Yuan, Adsorption of pairs of NOx molecules on singlewalled carbon nanotubes and formation of NO + NO3 from NO2, Surf. Sci. 603 (2009) 3234-3238. |
| [9] | Y.H. Zhang, K.G. Zhou, X.C. Gou, et al., Effects of dopant and defect on the adsorption of carbon monoxide on graphitic boron nitride sheet: a first-principles study, Chem. Phys. Lett. 484 (2010) 266-270. |
| [10] | J. Weimann, Toxicity of nitrous oxide, Best Pract. Res. Clin. Anaesthesiol. 17 (2003) 47-61. |
| [11] | M.W. Schmidt, K.K. Baldridge, J.A. Boatz, et al., General atomic and molecular electronic structure system, J. Comput. Chem. 14 (1993) 1347-1363. |
| [12] | N.M. O'Boyle, A.L. Tenderholt, K.M. Langner, cclib: a library for package-independent computational chemistry algorithms, J. Comput. Chem. 29 (2008) 839-845. |
| [13] | L.-H. Gan, J.-Q. Zhao, Theoretical investigation of [5,5], [9,0] and [10,10] closed SWCNTs, Physica E 41 (2009) 1249-1252. |
| [14] | J. Beheshtian, A.A. Peyghan, Z. Bagheri, Adsorption and dissociation of Cl2 molecule on ZnO nanocluster, Appl. Surf. Sci. 258 (2012) 8171-8176. |
| [15] | T.C. Dinadayalane, J.S. Murray, M.C. Concha, P. Politzer, J. Leszczynski, Reactivities of sites on (5,5) single-walled carbon nanotubes with and without a Stone-Wales defect, J. Chem. Theory Comp. 6 (2010) 1351-1357. |
| [16] | V. Nagarajan, R. Chandiramouli, NiO nanocone as a CO sensor: DFT investigation, Struct. Chem. 25 (2014) 1765-1771. |
| [17] | S. Tomic, B. Montanari, N.M. Harrison, The group III-V's semiconductor energy gaps predicted using the B3LYP hybrid functional, Physica E 40 (2008) 2125-2127. |
| [18] | S.F. Boys, F. Bernardi, The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys. 19 (1970) 553-566. |
| [19] | X.H. Deng, D.Y. Zhang, M.S. Si, M.S. Deng, The improvement of the adsorption abilities of some gas molecules on g-BN sheet by carbon doping, Physica E 44 (2011) 495-500. |
| [20] | M. Moradi, N2O reduction over hexagonal BN nanosheet: effects of Stone-Wales defect and carbon pair doping, Struct. Chem. 25 (2014) 1457-1463. |
| [21] | A. Ahmadi Peyghan, N.L. Hadipour, Z. Bagheri, Effects of Al doping and doubleantisite defect on the adsorption of HCN on a BC2N nanotube: density functional theory studies, J. Phys. Chem. C 117 (2013) 2427-2432. |
| [22] | M. Hjiri, L. El Mir, S.G. Leonardi, et al., Al-doped ZnO for highly sensitive CO gas sensors, Sens. Actuators B: Chem. 196 (2014) 413-420. |