




Combretastatinswere first extracted fromthe bark of the South African bush willow tree combretum caffrum in 1982 by Pettit et al. [1]. Those compounds may serve as a new anticancer drug that utilized starvation tactics to attack solid tumors. Among the combretastatin family,combretastatin A-4 (CA-4) (Fig. 1; (Z)-5- (3,4,5-trimethoxystyryl)-2-methoxyphenol) exerts a potent inhi- bition of tubulin polymerization by binding to the colchicine site and,as a consequence,demonstrates strong activity in suppressing tumor blood flow (TBF). Several studies described its ability to induce widespread necrosis of solid tumors,including multidrug- resistant ones,which suggested CA-4 is an attractive lead compound for the development of novel antitubulin anticancer agents [2, 3].

|
Download:
|
| Fig. 1.Structure of combretastatin A-4. | |
However,CA-4 does not show in vivo efficacy due to its poor pharmacokinetics resulting from its high lipophilicity,low aqueous solubility and also isomerization of cis-double bond to the more thermally stable trans-isomer [4, 5, 6]. So,considerable efforts have gone into modifying CA-4 to improve its water solubility and in vivo efficacy. To date,various CA-4 analogs have been synthesized and reported to possess cytotoxic activity against sever cancer cell lines. For example,a pro-drug of CA-4,the water- soluble phosphate derivative CA-4P (fosbretabulin) is nowin phase II clinical trials. Another combretastatin A-4 analog,the serine amide AVE8062 (ombrabulin) is not only in a phase II clinical trial in combination with taxanes and platinum salts in advanced solid tumors,but also in a phase II/III clinical trial in patients with advanced soft tissue sarcoma [7, 8, 9].
To study the relationship between the structure of CA-4 and the anti-proliferative effect in human cancer cells (SAR study),we synthesized 11 CA-4 analogs containing (i) hydrogenated deriva- tives,(ii) ethoxyl derivatives,(iii) amino derivatives and (iv) pro- drugs. The anti-proliferative effect of each class was tested using the MTT assay in human cancer cells in vitro. To understand the SAR of the CA-4 analogs,Surflex-Docking was applied to study the interactions between these analogs and tubulin. Furthermore,we have designed and synthesized a number of pro-drugs of potent CA-4 analogs to increase the water solubility.
2. Experimental 2.1. SynthesisAll the chemicals and reagentswere commercially available and required no further purifications. Solvents (THF,DMF,CH2Cl2, benzene) were dried and freshly distilled before use according to literature procedures. Chromatographic separations were per- formed on silica gel flash columns. TLC analyses were performed on precoated silica gel polyester plateswith a fluorescent indicator UV 254. Melting points were determined using a melting point apparatus (WRS-2A) and uncorrected. 1 H NMR and 13 C NMR spectra were recorded on a Bruker AVANCE III at 500 MHz or on a Bruker spectrometer at 101 Hz in chloroform-d using TMS (δ = 0.0 ppm) as an internal standard. IR was recorded on a NICOLET 6700 FT-IR. HRMS were recorded on a solanX 70 FT-MS spectrometer using methanol and water (v/v = 1:1) as solvent. LC- Mass spectra were recorded on a LCMS-2020 spectrometer from Shimadzu Corporation with acetonitrile and water as the mobile phase and the gradientwas from5% of acetonitrile at 0 min to 100% of acetonitrile at 10 min.
(Z)-2-Methoxy-5-(3,4,5-trimethoxystyryl)phenol (CA-4): Follow- ing the syntheticmethod fromShen et al. [10],compound CA-4was obtained in 35% yield. Mp 116.8-117.6 °C. 1 H NMR (CDCl3, 500 MHz): δ 3.69 (s,6H),3.84 (s,6H),3.85 (s,3H),5.58 (s,1H), 6.41 (d,1H,J = 12.0 Hz),6.46 (d,1H,J = 12.0 Hz),6.53 (s,2H),6.73 (d,1H,J = 8.5 Hz),6.79 (dd,1H,J = 8.5 Hz,J = 2.0 Hz),6.92(d,1H, J = 2.0 Hz).
2-Methoxy-5-(3,4,5-trimethoxyphenethyl)phenol (sit-1): A solu- tion of compound CA-4 (1.0 g,3.16 mmol) and palladium-carbon (10%,0.1 g) in MeOH (20 mL) was stirred at room temperature for 2 h under hydrogen atmosphere. Palladium-carbon was filtered off,the filtrate was dried over Na2SO4 and concentrated. Compound sit-1 was obtained in 91% yield. 1 H NMR (500 MHz, CDCl3): d 6.81 (d,1H,J = 3.0 Hz),6.77 (d,1H,J = 8.0 Hz),6.64-6.66 (m,1H),6.38 (s,2H),5.61(s,1H),3.87 (s,3H),3.83 (s,9H), 2.82 (s,4H).
5-(3-(Benzyloxy)-4-ethoxystyryl)-1,2,3-trimethoxybenzene (sit- 11) (Scheme 1): A mixture of 4-hydroxy-3-methoxybenzaldehyde 1 (1 g,6.57 mmol) and potassiumcarbonate (1.36 g,9.85 mmol) in DMF (20 mL) was stirred at 40 °C for 10 min. Ethyl bromide (0.98 mL,13.1 mmol) was added through the septum after the mixture was raised to 70 °C. The reaction mixture was then stirred at this temperature until TLC analysis indicated the completion of the reaction,then quenched by water,extracted with EtOAc,dried over Na2SO4 and concentrated. Pure white 4-ethoxy-3-methox- ybenzaldehyde 2 was obtained by crystallization (petroleumether (60-90 °C): EtOAc = 9:1,v/v). mp 62.1-62.2 °C. 1 H NMR (CDCl3, 500 MHz): δ 9.84 (s,1H),7.41-7.45 (m,2H),6.96 (d,1H,J = 5.0 Hz), 5.80 (s,1H),4.19 (q,2H,J = 5.0 Hz),3.93 (s,3H),1.50 (t,3H, J = 5.0 Hz).

|
Download:
|
| Scheme.1.Synthesis of sit-2,sit-8,sit-11 by our group before. | |
A solution of 4-ethoxy-3-methoxybenzaldehyde 2 (1 g, 5.6 mmol),p-toluenesulfonic acid (60 mg,0.31 mmol) and ethyl- ene glycol (6 mL,0.1 mol) in benzene (30 mL) was refluxed for 12 h. After cooling to room temperature,aqueous potassium carbonate (15%,25 mL)was added. Organic layerwaswashedwith aqueous potassiumcarbonate (15%,50 mL),dried over Na2SO4 and concentrated,white 2-(4-ethoxy-3-methoxyphenyl)-1,3-dioxo- lane 3 was obtained. mp 75-77 °C. 1 H NMR (CDCl3,500 MHz): δ 7.21 (s,1H),6.83 (d,2H,J = 5.0 Hz),5.80 (s,1H),4.00 (t,4H, J = 5.0 Hz),4.04 (q,2H,J = 5.0 Hz),3.83 (s,3H),1.43 (t,3H, J = 5.0 Hz).
A solution of diphenylphosphine (1 mL,5.8 mmol) and n-butyl lithium(2.5 mol/L in hexanes,3 mL) in anhydrous THF (10 mL)was stirred in an ice bath under nitrogen atmosphere. 2-(4-Ethoxy-3- methoxyphenyl)-1,3-dioxolan 3 (1 g,4.4 mmol) was dissolved in anhydrous THF (5 mL) and added. Then the solution was stirred at room temperature until TLC analysis indicated the completion of the reaction. The mixture was quenched with water. Aqueous phase was acidified with HCl when the yellowmixture became tea green and the product was extracted with EtOAc,dried over Na2SO4 and evaporated in vacuum. Pure white 4-ethoxy-3- hydroxybenzaldehyde 5 was obtained after column chromatogra- phy purification (petroleum ether (60-90 °C): EtOAc = 5:1,v/v). mp 125-127 °C. 1 H NMR (CDCl3,500 MHz): δ 9.84 (s,1H),7.41- 7.44 (m,2H),6.94 (d,1H,J = 10.0 Hz),5.80 (s,1H),4.22 (q,2H, J = 5.0 Hz),1.50 (t,3H,J = 5.0 Hz).
A mixture of 4-ethoxy-3-hydroxybenzaldehyde 5 (1 g, 6.02 mmol) and potassium carbonate (1.24 g,9 mmol) in EtOH (20 mL) was stirred at 40 °C for 10 min. Benzyl chloride (0.7 mL, 6.1 mmol) was added through the septum. Then the reaction mixture was refluxed for 3 h. After cooling to 50 °C,the solution was filtered. Pure white 3-(benzyloxy)-4-ethoxybenzaldehyde 6 was obtained after the filtrate was cooled in a fridge. mp 69.8- 70.8 °C. 1 H NMR (CDCl3,500 MHz): δ 9.81 (s,1H),6.98-7.47 (m, 8H),5.20 (s,2H),4.19 (q,2H,J = 5.0 Hz),1.50 (t,3H,J = 5.0 Hz). 13 C NMR (CDCl3,101 MHz): δ 190.8,154.7,148.8,136.6,129.8,128.5, 127.9,127.2,126.8,112.3,111.9,71.0,64.6,14.6. MS (m/z): 257 (M+ ). ESI-HRMS (m/z): calcd. for C16H17O3 (M+H) + ,257.1178. Found: 257.1196. IR (KBr) (vmax,cm-1 ): 2985,2935,2820,1686, 1595,1580,1436,1278,1266,1131,1001,874,802,750.
NaH (60%,0.64 g,26.6 mmol) was added to anhydrous DMSO (10 mL) and themixturewas stirred at roomtemperature (Scheme 2). A solution of 3,4-dihydroxybenzaldehyde 9 (1 g,7.24 mmol) in anhydrous DMSO (5 mL) was added dropwise through a syringe. The reaction mixture was stirred for about half an hour. Benzyl bromine (0.86 mL,7.3 mmol) was then added dropwise by a syringe and the resulting solution was stirred overnight. The mixture was neutralized with HCl (2 mol/L) and extracted with EtOAc,dried over Na2SO4 and evaporated in vacuum. Pure 3- (benzyloxy)-4-hydroxybenzaldehyde 10 was obtained after col- umn chromatography purification (petroleum ether (60-90 °C): EtOAc = 3:1,v/v). mp 114.3-114.7 °C. 1 H NMR (CDCl3,500 MHz): δ 9.82 (s,1H),7.51 (d,1H,J = 5.0 Hz),7.40-7.46 (m,6H),7.06 (d,1H, J = 10.0 Hz),6.26 (s,1H),5.18 (s,2H).

|
Download:
|
| Scheme.2.New method for the synthesis of sit-2 and sit-11. | |
3-(Benzyloxy)-4-hydroxybenzaldehyde 10 (1 g,4.38 mmol) and potassium carbonate (0.91 g,6.57 mmol) in DMF (15 mL) was stirred at 40 °C for 10 min. Ethyl bromide (0.65 mL, 12.0 mmol) was added through the septum after the mixture was raised to 70 °C. The reaction mixture was then stirred at this temperature until TLC analysis indicated the completion of the reaction,then the mixture was quenched by water,extracted with EtOAc,dried over Na2SO4 and concentrated. Pure white 3- (benzyloxy)-4-ethoxybenzaldehyde 6 was obtained by crystalliza- tion (petroleum ether (60-90 °C): EtOAc = 9:1,v/v).
Compound 7 (4 g,7.66 mmol) was dissolved in anhydrous THF (40 mL),and the mixture was stirred in an ice bath under nitrogen atmosphere,t-BuOK (1.31 g,11.7 mmol) was added,then 3- (benzyloxy)-4-ethoxybenzaldehyde 6 (1 g,3.90 mmol) was dis- solved in anhydrous THF (10 mL) and added through the septum. The mixture was stirred at room temperature for 4 h. TLC analysis indicated the completion of the reaction,then the mixture was quenched by water,extracted with EtOAc,dried over Na2SO4 and concentrated under reduced pressure. Pure compound sit-11 was obtained in 78% yield after column chromatography purification (petroleum ether (60-90 °C): EtOAc = 10:1,v/v). mp 135.8-136 °C.1 H NMR (CDCl3,500 MHz): δ 7.49 (d,2H,J = 5.0 Hz),7.37-7.41 (m, 2H),7.32-7.33 (m,1H),7.12 (d,1H,J = 5.0 Hz),7.05-7.07 (m,1H), 6.81-6.93 (m,3H),6.70 (s,2H),5.19 (s,2H),4.13 (q,2H,J = 5.0 Hz), 3.91 (s,6H),3.86 (s,3H),1.46 (t,3H,J = 5.0 Hz). 13 C NMR (CDCl3, 101 MHz): δ 153.4,149.2,148.7,137.7,137.3,133.3,130.4,128.5, 127.9,127.8,127.3,126.8,120.5,113.7,112.9,103.3,71.5,64.6, 60.9,56.1,14.9; MS (m/z): 421 (M+ ); ESI-HRMS (m/z): Calcd. for C26H29O5 (M+H) + : 421.2015,Found: 421.2043; IR (KBr) (vmax, cm-1 ): 2966,2934,2835,2357,1581,1508,1274,1242,1130.
2-Ethoxy-5-(3,4,5-trimethoxyphenethyl)phenol (sit-2): A mix- ture of compound sit-11 (1.0 g,2.38 mmol) and palladium-carbon (10%,0.2 g) in MeOH (25 mL) was stirred at room temperature for 3 h under hydrogen atmosphere. Palladium-carbon was filtered, the filtrate was dried over Na2SO4 and concentrated. Compound sit-2 was obtained in 92% yield. mp 69.3-69.6 °C. 1 H NMR (CDCl3, 500 MHz): δ 6.81 (s,1H),6.75 (d,1H,J = 5.0 Hz),6.62 (d,1H,J = 5.0 Hz),6.38 (s,2H),5.63 (s,1H),4.09 (q,2H,J = 5.0 Hz),3.83 (s, 9H),2.82 (s,4H),1.43 (t,3H,J = 5.0 Hz). 13 C NMR (CDCl3,101 MHz): d 153.0,145.6,144.0,137.6,136.2,134.9,119.7,114.6,111.5, 105.4,64.6,60.8,56.0,38.4,37.3,14.9; MS (m/z): 333 (M+ ); ESI- HRMS (m/z): Calcd. for C19H25O5 (M+H) + : 333.1702; Found: 333.1741; IR (KBr) (vmax,cm-1 ): 3345,2975,2930,2867,1592, 1526,1464,1425,1247,1115,1003,869.
2-Ethoxy-5-(3,4,5-trimethoxyphenethyl)phenyl sodiumphosphate (sit-8): A solution of phosphorus oxychloride (0.8 mL,9.0 mmol) in CH2Cl2 (3 mL) was stirred in an ice bath. Compound sit-2 (1 g, 3.01 mmol) was dissolved in CH2Cl2 (5 mL) and added. A solution of triethylamine (1.87 mL,13.5 mmol) in CH2Cl2 (3 mL) was added dropwise after the mixture was stirring for 5 min. TLC analysis indicated the completion of the reaction,then the mixture was quenched by water,extracted with CH2Cl2,dried over Na2SO4 and concentrated. The viscous oilwas cooled in an ice bath,neutralized to 8-10 (pH) with 2 mol/L sodium hydroxide and the mixture was stirred for 8 h at 70 °C,then filteredwhile hot,the filtratewas dried over Na2SO4 and concentrated. Pure white compound sit-8 was obtained in 50% yield by re-crystallization from hot EtOH. mp 140.3-141.0 °C. 1 H NMR (D2O,500 MHz): δ 7.38 (s,1H),6.84 (d,1H, J = 10.0 Hz),6.65 (d,1H,J = 5.0 Hz),6.54 (s,2H),4.04 (q,2H, J = 5.0 Hz),3.74 (s,6H),3.67 (s,3H),2.78-2.81 (m,4H),1.33 (t,3H, J = 5.0 Hz). 13 C NMR (D2O,101 MHz): δ 152.1,146.7,143.7,139.0, 135.1,134.7,121.7,120.3,114.2,105.9,65.4,60.7,55.8,48.9,37.4, 36.5,14.1. 31 P NMR (D2O,500 MHz): δ 3.20(s). MS (m/z): 456 (M+ ). ESI-HRMS (m/z): calculated for C19H23Na2O8P (M+Na) + : 479.0824, found: 479.0813. IR (KBr) (vmax,cm-1 ): 3242,2599,2239,1982, 1589,1505,1092,988.
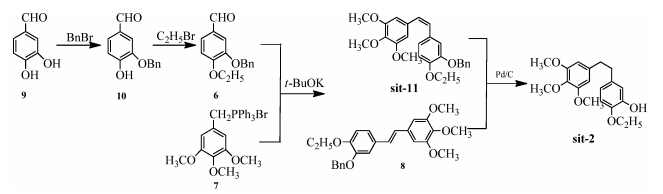
(Z)-5-(4-Ethoxy-3-nitrostyryl)-1,2,3-trimethoxybenzene (sit-6): A mixture of 4-hydroxy-3-nitrobenzaldehyde 11 (1 g,6.0 mmol) and potassium carbonate (1.24 g,9 mmol) in DMF (15 mL) was stirred at 40 °C for 10 min. Ethyl bromide (0.9 mL,12.0 mmol)was added through the septum after the mixture was raised to 70 °C. The reaction mixture was then stirred at this temperature until TLC analysis indicated the completion of the reaction,then the mixturewas quenched bywater,extractedwith EtOAc,dried over Na2SO4 and concentrated. Pure white 4-ethoxy-3-nitrobenzalde- hyde 12was obtained by re-crystallization (petroleumether (60- 90 °C): EtOAc = 9:1,v/v). mp 46.4-46.9 °C. 1 H NMR (CDCl3, 500 MHz): δ 9.93 (s,1H),8.33 (s,1H),8.06-8.08 (m,1H),7.21 (d,1H,J = 10.0 Hz),4.30 (q,2H,J =5.0Hz),1.53(t,3H,J =5.0Hz). Compound 7 (4 g,7.66 mmol) was dissolved in anhydrous THF (40 mL),and themixturewas stirred in an ice bath under nitrogen atmosphere,n-butyl lithium (2.5 mol/L in hexanes,2.6 mL) was added,then 4-ethoxy-3-nitrobenzaldehyde 12 (1 g,5.13 mmol) was dissolved in anhydrous THF (10 mL) and added through the septum. The mixture was stirred at room temperature for 12 h. TLC analysis indicated the completion of the reaction,then the mixture was quenched by water,extracted with EtOAc,dried over Na2SO4 and concentrated under reduced pressure. Pure compound sit-6 was obtained in 50% yield after column chroma- tography purification (petroleumether (60-90 °C): EtOAc = 10:1,v/ v). mp 101.8-102.4 °C. 1 H NMR (CDCl3,500 MHz): δ 7.77 (s,1H), 7.39-7.41 (m,1H),6.92 (d,1H,J = 10.0 Hz),6.57 (d,1H,J =10.0Hz), 6.43-6.47 (m,3H),4.15 (q,2H,J = 5.0 Hz),3.85 (s,3H),3.72 (s,6H), 1.46 (t,3H,J = 5.0 Hz). 13 C NMR (CDCl3,101 MHz): δ 153.2,151.0, 139.9,137.7,134.4,131.8,131.2,129.5,126.9,125.8,114.1,105.9, 65.5,61.0,56.0,14.5.MS (m/z):359(M+ ).ESI-HRMS (m/z): calculated for C19H22NO6 (M+H) + : 360.1447,found: 360.1438. IR (KBr) (vmax, cm-1 ): 2957,2835,1735,1621,1577,1530,1503,1458,1429,1428, 1412,1349,1333,1164,1129,1037,1005,968,930,856.
2-Ethoxy-5-(3,4,5-trimethoxyphenethyl)benzenamine (sit-3):A solution of compound sit-6 (1.0 g,2.79 mmol) and palladium- carbon (10%,0.2 g) in MeOH/EtOAc (25 mL,1:1.5,v/v) was stirred at room temperature for 3 h under hydrogen atmosphere. Palladium-carbon was filtered off,the filtrate was dried over Na2SO4 and concentrated. Compound sit-3 was obtained in 90% yield. mp 82.9-83.4 °C. 1 H NMR (CDCl3,500 MHz): δ 6.70 (d,1H, J = 5.0 Hz),6.59 (d,1H,J = 5.0 Hz),6.51-6.53 (m,1H),6.39 (s,2H), 4.04 (q,2H,J = 5.0 Hz),3.83 (s,9H),2.75-2.83 (m,4H),1.42 (t,3H, J = 5.0 Hz). 13 C NMR (CDCl3,101 MHz): δ 153.1,146.7,145.9,137.6, 136.2,135.2,134.4,128.1,122.7,119.7,111.9,64.3,60.9,56.1,38.5, 37.2. MS (m/z): 331 (M+ ). ESI-HRMS (m/z): calculated for C19H25NaNO4 (M+Na) + ,354.1681,found: 354.1691. IR (KBr) (vmax, cm-1 ): 3434,3350,3050,2986,2978,2934,2837,1620,1587, 1517,1507,1474,1455,1421,1392,1325,1290,1237,1226,1179, 1128,1048,1021,1007,973,952,921,843.
2-Amino-N-(2-ethoxy-5-(3,4,5-trimethoxyphenethyl)phenyl)-3- hydroxypropanamide (sit-9): A mixture of compound sit-3 (1 g, 3.02 mmol),Fmoc-L-serine (1.03 g,3.15 mmol),DCC (0.65 g, 3.15 mmol),HOBT (0.43 g,3.15 mmol) in DMF (20 mL) was stirred at room temperature for 5 h under nitrogen atmosphere. TLC analysis indicated the completion of the reaction,then themixture was diluted by EtOAc (10 mL),dried over Na2SO4 and concentrated under reduced pressure. The crude product was dissolved in CH2Cl2/MeOH (1:1,10 mL),NaOH solution (2.5 mL,2 mol/L) was added. The reaction mixture was stirred at room temperature for 24 h. After cooling,the solvents was quenched by saturated salt water,extracted with CH2Cl2,dried over Na2SO4 and concentrated. The residue was purified by column chromatography (petroleum ether (60-90 °C): EtOAc = 1:2,v/v) to afford the compound sit-9 in 45% yield. mp 162.0-162.5 °C. 1 H NMR (CDCl3,500 MHz): δ 9.92 (s,1H),8.29 (s,1H),6.82-6.83 (m,1H),6.78 (d,1H,J = 5.0 Hz),6.40 (s,2H),4.07(q,2H,J = 5.0 Hz),4.03 (q,1H,J = 5.0 Hz),3.88 (q,1H, J = 5.0 Hz),3.83 (d,9H,J = 5.0 Hz),3.73 (s,1H),2.83 (s,4H),2.57 (s, 2H),1.44 (t,3H,J = 5.0 Hz). 13 C NMR (CDCl3,101 MHz): δ 171.2, 153.0,146.2,137.7,136.2,134.3,127.2,123.9,119.7,111.2,105.5, 100.0,64.5,60.8,56.6,56.1,38.6,37.6,29.7,14.9. MS (m/z): 418 (M+ ). ESI-HRMS (m/z): calculated for C22H31N2O6 (M+H) + , 419.2182,found: 419.2170. IR (KBr) (vmax,cm-1 ): 3366,3283, 2927,2012,1670,1593,1552,1507,1473,1386,1329,1290,1230, 1185,1127,1057,1011,972,890,871,854.
2-Amino-N-(2-ethoxy-5-(3,4,5-trimethoxyphenethyl)phenyl)ace- tamide (sit-10): A mixture of compound sit-3 (1 g,3.02 mmol), Fmoc-glycine (0.94 g,3.15 mmol),BOP (1.40 g,3.15 mmol) in DMF (20 mL)was stirred at 60 °C for 2 h under nitrogen atmosphere. TLC analysis indicated the completion of the reaction,then themixture was quenched by saturated NaHCO3 (20 mL),extracted with CH2Cl2,dried over Na2SO4 and concentrated under reduced pressure. The crude product was dissolved in MeOH (15 mL) and NaOH solution (2.5 mL,2 mol/L) was added. The reaction mixture was stirred at room temperature for 3 h until the TLC analysis indicated the completion of the reaction. After cooling,the solvents was quenched by saturated NaCl solution,extracted with CH2Cl2,dried over Na2SO4 and concentrated. The residue was purified by column chromatography (petroleum ether (60-90 °C):EtOAc = 1:1,v/v) to afford the compound sit-10 in 50% yield. mp 92.1-93.9 °C. 1 H NMR (CDCl3,500 MHz): δ 9.79 (s,1H),8.38 (s,1H), 6.77-6.84 (m,2H),6.42 (s,2H),4.08 (q,2H,J = 10.0 Hz),3.84 (d,9H, J = 5.0 Hz),3.56 (s,1H),2.84 (s,4H),2.19 (s,2H),1.45 (t,3H, J = 10.0 Hz). 13 C NMR (CDCl3,101 MHz): δ 170.4,153.0,146.0, 137.8,136.1,134.4,127.6,123.5,119.5,111.1,100.0,64.4,60.9, 56.1,45.5,38.7,37.7,14.9. MS (m/z): 388 (M+ ). ESI-HRMS (m/z): calcd. for C21H29N2O5 (M+H) + ,389.2076,found: 389.2076. IR (KBr) (vmax,cm-1 ): 3402,3271,2998,2933,2835,1674,1592,1540, 1458,1366,1334,1289,1254,1235,1183,1130,1079,1037,1004, 976,921,859.
(Z)-1,2,3-Trimethoxy-5-(4-methoxy-3-nitrostyryl) benzene (sit- 7): Following the synthetic method from Pettit et al. [11], compound sit-7 was obtained in 25% yield. 1 H NMR (500 MHz, CDCl3): d 8.03 (s,1H),7.68 (d,1H J = 5 Hz),7.11 (d,1H,J = 5 Hz), 7.00 (q,2H,J = 5 Hz),6.75 (s,2H),4.01(s,3H),3.94 (s,6H),3.90 (s, 3H).
2-Methoxy-5-(3,4,5-trimethoxyphenethyl)benzenamine (sit-4):A solution of compound sit-7 (1.0 g,2.90 mmol) and palladium- carbon (10%,0.2 g) in MeOH/EtOAc (25 mL,1:1.5,v/v) was stirred at roomtemperature for 3 h. Palladium-carbonwas filtered off,the filtrate was dried over Na2SO4 and concentrated. Compound sit-4 was obtained in 90% yield. 1 H NMR (500 MHz,CDCl3),d 6.71 (d,1H, J = 10 Hz),6.58 (d,1H,J = 5 Hz),6.55 (d,1H,J = 10 Hz),6.39 (s,2H), 3.84 (s,12H),2.80 (s,4H),1.58(s,2H). (Z)-2-Methoxy-5-(3,4,5-trimethoxystyryl)benzenamine (sit-5):A solution of compound sit-7 (1.0 g,2.90 mmol) and zinc powder (1.89 g,29 mmol) in AcOH (30 mL) was stirred at room tempera- ture for 6 h. Themixturewas filtered and the filtratewas dried over Na2SO4 and concentrated. The residue was purified from crystalli- zation (petroleum ether (60-90 °C): EtOAc = 9:1,v/v) to afford compound sit-5 in 73% yield. 1 H NMR (500 MHz,CDCl3): d 6.95 (s, 1H),6.90 (d,3H,J = 10 Hz),6.80 (d,1H,J = 5 Hz),6.72 (s,2H),3.90 (s,12H).
2.2. Cell growth conditions and anti-proliferative assayTo better characterize drug-induced cytotoxicity of these compounds (sit-1 to sit-11) in contrast with CA-4,some human cancer cells like human hepatocellular liver carcinoma cell (HepG2),human cholangiocarcinoma (QBC939),human breast cancer cells (SK-BR-3),human colon cancer cell (HCT-8) and human gastric cancer cell (MKN45) obtained from the Cell Bank of Chinese Academy of Science were treated. All of these cells were maintained in RPMI 1640 medium with 10% fetal bovine serum at 37 °C in a humidified atmosphere with 5% CO2. All cells were seeded into 96-well flat-bottomed culture plates in triplicates separately with 10 mg/mL WA or GsA for 44 h. 500 mg/mL 3-(4,5- dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide (MTT) was added and cells were then incubated for another 4 h. The IC50 values were calculated through the determination of lactate dehydrogenase (LDH) in cell culture supernatant using GraphPad Prism5.0 software.
2.3. Molecular modeling
Energy minimization: Minimum energy conformations of all 11
CA-4 analogs and CA-4 were calculated using theminimizemodule
of Sybyl-X2.0 [12]. The forcefieldwas calculatedwithMMFF94 at an
8A ˚
cutoff fornon-bonded interactions,and the atomic point charges
were also calculated using MMFF94. Minimizations were achieved
using the consecutive steepest descent method for the first 100
steps,conjugate gradient (Powell) and quasi-Newton (BFGS; named
for its originators,and approximates the inverse of the Hessian
matrix) energy minimization steps until the root-mean-square
(RMS) of the gradient became less than 0.005 kcal mol-1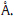 .
.
Docking calculations: The Surflex-Dock [13] module imple- mented in the Sybyl program was used for the docking studies. All CA-4 analogs were docked into a tubulin crystal structure (PDB ID: 3UT5) by an empirical scoring function and a patented search engine in Surflex-Dock. Protomol,a representation of a ligand making every potential interaction with the binding site,was applied to guide molecular docking. Protomols could be estab- lished by three manners: (1) Automatic: Surflex-Dock finds the largest cavity in the receptor protein; (2) Ligand: a ligand in the same coordinate space is used as the receptor; (3) Residues: residues in the receptor are specified [13]. In this study,the automatic docking was applied. Other parameters were estab- lished by default in the software. Surflex-Dock scores (total scores) were expressed in -lgKd units to represent binding affinity.
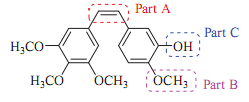
3. Results and discussion 3.1. ChemistryThe synthesis of diphenylethane derivates (sit-2,sit-3,sit-6, sit-8,sit-9,sit-10,sit-11) have been reported by our group before (Scheme 1) [14]. For the construction of this series of dipheny- lethane derivatives,the synthesis of 3-(benzyloxy)-4-ethoxyben- zaldehyde 6 was the main challenge. Firstly,the synthetic sequence was long. Secondly,the use of expensive,unstable and toxicmetal reducers and the need for slowincrease of temperature fromice bath to roomtemperatures in the procedures have limited the product yields. With our continuing interesting in preparing various CA-4 derivatives,we herein demonstrate a simpler route for the preparation of various diphenylethane derivates with 3,4- dihydroxybenzaldehyde 9 using theWittig reaction as a key step as illustrated in Scheme 2. 3-(Benzyloxy)-4-hydroxybenzaldehyde 6 was obtained by reaction of commercially material 3,4-dihydrox- ybenzaldehyde 9 with benzyl bromide [15]. Subsequent alkylation of the intermediate aldehyde 10 gave 3-(benzyloxy)-4-ethoxy- benzaldehyde 6.
The modified synthesis of 3-(benzyloxy)-4-hydroxybenzalde- hyde 6 required only 2 steps compared to 5 steps in the original method. And the total yield of compound 6 increased from 30% to 63%. A simple,facile,high yield,less cumbersome and environ- mental friendly synthesis of 3-(benzyloxy)-4-ethoxybenzaldehyde 6 was established.
The aldehyde 6 was reacted with triphenyl(3,4,5-trimethox- ybenzyl)phosphonium bromine 7 and potassium tert-butoxide (t- BuOK) to furnish the Wittig product 5-(3-(benzyloxy)-4-ethox- yphenethyl)-1,2,3-trimethoxybenzene sit-11. Diphenylethene sit- 11 was hydrogenated under hydrogen balloon conditions using palladium-carbon as a catalyst to afford the final product sit-2 in an overall yield of 43%.
The synthesis of the amino derivatives of sit-3,sit-6,sit-9 and sit-10 are outlined in Scheme 3. 4-Ethoxy-3-nitrobenzaldehyde 12 was obtained from the alkylation of commercial compound 4- hydroxy-3-nitrobenzaldehyde 11. Then sit-6 was prepared from 12 via theWittig reaction. sit-3 was easy to synthesis following the general method. DCC and BOP were used to promote the coupling of sit-3 and Fmoc-L-Ser or Fmoc-Gly to produce b>sit-9 and sit-10.

|
Download:
|
| Scheme.3.Synthesis of sit-3,sit-9 and sit-10. | |
The MTT assay was carried out to investigate the anti- proliferative activity of all synthesized CA-4 analogs (sit-1 to sit-11) in human cervix carcinoma (HeLa). CA-4 was used as a positive control in the assay. The IC50 values for sit-1,sit-2,sit-3, sit-4 and sit-8 were 0.22 μmol/L,0.11 μmol/L,0.20 μmol/L, 0.16 μmol/L and 0.57 μmol/L,respectively (Table 1). This result demonstrated that analogs sit-1,sit-2,sit-3 and sit-4 have significant effects in vitro,when comparedwith the lead compound CA-4 (the IC50 value of CA-4 is 0.40 mmol/L). The anti-proliferative activities of sit-1,sit-2 and sit-3 were also tested in human hepatocellular liver carcinoma cell (HepG2),human cholangio- carcinoma (QBC939),human breast cancer cells (SK-BR-3),human colon cancer cell (HCT-8) and human gastric cancer cell (MKN45). The IC50 value of each analog shows that they all have some effect on each cancer cell in vitro,as shown in Table 2.
| Table 1 Structure,purity,yield and in vitro antiproliferative activity of CA-4 analogs on HeLa. |

The double bond in Part A of CA-4 was hydrogenated to afford the final product sit-1. The anti-proliferative effect of sit-1 was 2- fold more than that of CA-4,which suggested that the single bond is better than the double bond in Part A for the cytotoxicity in human cancer cells. The IC50 value for sit-4 (a diphenylethane derivative) was 0.16 mmol/L,but sit-5 (a diphenylethene deriva- tive) has no effect in HeLa cells. This result further validated the above conclusion.
Themethoxy group in Part B of sit-1 was replaced by an ethoxy group to afford sit-2. The IC50 value of sit-2 for each human cancer cellswas near half of that of sit-1 in Table 2. It suggested the ethoxy group is better than themethoxy group for anti-proliferative effect.
The Part C in sit-2 is a hydroxyl group,and in sit-3 is an amino group. Both compounds have some effect in those six human cancer cells (Table 2). The anti-proliferative activities of sit-2 and sit-3 in HepG2,SK-BR-3,HCT-8 are similar. The anti-proliferative effect of sit-2 was 2-fold more than that of sit-3 in HeLa and QBC939. There is no significant difference between hydroxyl and amino groups in Part C for the anti-proliferative effect. But the nitro group (sit-6 and sit-7) or benzyloxy group (sit-11) can abolish the anti-proliferative effect (Table 1).
| Table 2 Cytotoxicity against five cancer cell lines by compounds sit-1,sit-2 and sit-3. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
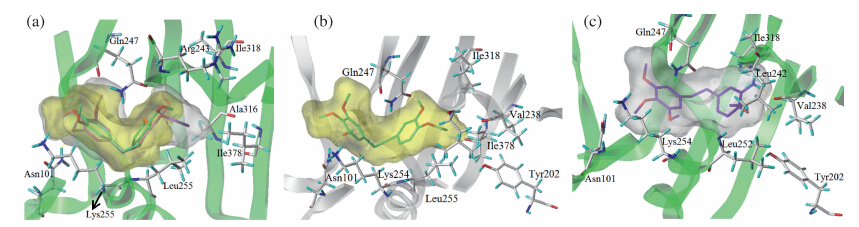
The colchicine binding pocket was used in the study of binding affinities of our analogs. The total score (a default scoring function in Sybyl) is an indication of the binding affinity of a ligand to its receptor. The scores of CA-4 and sit-1 are similar with values of 7.04 and 6.96,respectively. And their anti-proliferative effects are also similar. The amino acids residues around the Part B in CA-4 are hydrophobic residues,which suggested the bioactivity can be increased if the hydrophobicity of Part B in these analogs was enhanced (Fig. 2). This docking result makes a clear explanation of why the ethyoxyl is better than the methoxyl group for anti- proliferative effect. Both the hydroxyl and the amino groups in Part C can form one hydrogen bond with the receptor (Fig. 2),but the nitro group or benzyloxy group cannot form any hydrogen bond with the receptor. It also can explain the difference of the anti- proliferative effect of analogs sit-2,sit-3,sit-6,sit-7 and sit-11.

|
Download:
|
| Fig. 2.Fig. 2. Binding conformations of the analogs at the active site of tubulin. (a) Bindingmodel of sit-1 (methoxy derivative,itsmolecular surface was shown in yellow) and sit-2 (ethoxy derivative,its molecular surface was shown in white). This docking result makes a clear explanation of why the ethoxy is better than the methoxy group for anti- proliferative effect. (b) and (c) are binding structures of sit-2 (hydroxyl derivative) and sit-3 (amino derivative),respectively. Both hydroxyl derivative and amino derivative have only one hydrogen bond with the receptor (Asn101). | |
{kind=link}
As CA-4 does not show in vivo efficacy due to its poor water solubility,compound sit-8,sit-9 and sit-10 were synthesized as the pro-drugs of sit-2. The Clog P values (calculated using Sybyl) of these analogs showed the water-soluble phosphate or amino acid derivative can improve thewater solubility (data shown in Table 1).
In this paper,we designed and synthesized 11 CA-4 analogs, tested the anti-proliferative effect of each analog,and studied the structure-activity relationship of those analogs. These results will be useful for the design of new CA-4 analogs that are structurally related to those used in the current SAR study.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21472126,21402122) and Shanghai Municipal Natural Science Foundation (No. 12ZR450200).
| [1] | G.R. Pettit, G.M. Cragg, D.L. Herald, J.M. Schmidt, P. Lohavanijaya, Isolation and structure of combretastatin, Can. J. Chem. 60 (1982) 1374-1376. |
| [2] | G.R. Pettit, S.B. Singh, E. Hamel, et al., Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4, Experientia 45 (1989) 209-211. |
| [3] | A.A.E. El-Zayat, D. Degen, S. Drabek, et al., In vitro evaluation of the antineoplastic activity of combretastatin A-4, a natural product from Combretum caffrum (arid shrub), Anticancer Drugs 4 (1993) 19-25. |
| [4] | G.R. Pettit, B.E. Toki, D.L. Herald, et al., Antineoplastic agents. 410. Asymmetric hydroxylation of trans-combretastatin A-4, J. Med. Chem. 42 (1999) 1459-1465. |
| [5] | G.R. Pettit, M.R. Rhodes, D.L. Herald, et al., Antineoplastic agents 393. Synthesis of the trans-isomer of combretastatin A-4 prodrug, Anti-Cancer Drug Des. 13 (1999) 981-993. |
| [6] | K. Ohsumi, R. Nakagawa, Y. Fukuda, et al., Novel combretastatin analogues effective against murine solid tumors: design and structure-activity relationships, J. Med. Chem. 41 (1998) 3022-3032. |
| [7] | E.D. Durrant, J. Richards, A. Tripathi, et al., Development of water soluble derivatives of cis-3 4',5-trimethoxy-3'-aminostilbene for optimization and use in cancer therapy, Invest. New Drugs 27 (2009) 41-52. |
| [8] | P. Tresca, D. Tosi, L. Doorn, et al., Phase I and pharmacologic study of the vascular disrupting agent ombrabulin (Ob) combined with docetaxel (D) in patients (pts) with advanced solid tumors, J. Clin. Oncol. 28 (2010) 3023. |
| [9] | J.H. Jiang, C.H. Zheng, C.Q. Wang, et al., Synthesis and biological evaluation of 5,6,7-trimethoxy-1-benzylidene-3,4-dihydro-naphthalen-2-one as tubulin-polymerization inhibitors, Chin. Chem. Lett. (2015), http://dx.doi.org/10.1016/ j.cclet.2015.03.022. |
| [10] | W.P. Shen, Y.J. Diao, H.M. Jin, J.P. Wang, J.G. Wang[32_TD$DIF], Polymer-supported preparation of Combretastatin A4 using poly(4-(chlorodiphenylmethyl)styrene) as solid carrier, CN1704393A, 2005. |
| [11] | G.R. Pettit, M.R. Rhodes, D.L. Herald, et al., Antineoplastic agents. 445. Synthesis and evaluation of structural modifications of (Z)-and (E)-combretastatin A-4, J. Med. Chem. 48 (2005) 4087-4099. |
| [12] | Z.P. Kai, Y. Ling, W.J. Liu, F. Zhao, X.L. Yang, The study of solution conformation of allatostatins by 2-D NMR and molecular modeling, Biochim. Biophys. Acta 1764 (2006) 70-75. |
| [13] | R. Spitzer, A.N. Jain, Surflex-Dock: docking benchmarks and real-world application, J. Comput. Aided Mol. Des. 26 (2012) 687-699. |
| [14] | F.H. Wu, W.G. Zhou, F.M. Xu, F.H. Xiao, Ethoxy diphenyl ethane derivates, preparation processes and medical application as tubulin polymerization inhibitor, CN101723813A[34_TD$DIF], 2010. |
| [15] | O. Taratula, P.A. Hill, Y.B. Bai, N.S. Khan, I. Dmochowski, Shorter synthesis of trifunctionalized cryptophane-A derivatives, Org. Lett. 13 (2011) 1414-1417. |