




Multi-component reactions (MCR) have emerged as a powerful strategy in the preparation of heterocyclic compounds due to the advantages compared to conventional linear-type multi-step synthesis [5]. Major advantages of MCR include lower cost,shorter reaction time and overcoming time consuming and sparing of purification process. It is established that MCRs are in general much more environment friendly and offer rapid access to large compound librarieswith diverse functionalities [3]. The quinazolinonemoiety is a building block for many naturally occurring alkaloids [6, 7] such as chloroqualone [8],deoxyvasicinone [9],rutaecarpine [10] and bioactive molecules like methaqualone [19] (Fig. 1).

|
Download:
|
| Fig. 1.Representative examples of bioactive compounds containing quinazolinone motif. | |
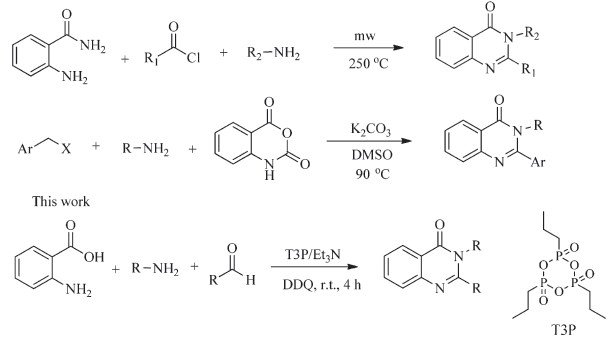
Quinazolinone alkaloids and substituted quinazolinones are a class of natural drugswhich display a variety of phormocological and therapeutic activities [20, 21],such as anti-inflamatory,anticonvulsant, antiulcer and antimalerial. Number of synthetic routes have been developed for the synthesis of 4(3H)-quinazolinones,which include copper-catalyzed N-arylation of o-bromobenzoic acid derivatives with amidines and subsequent intramolecular condensation affording 2 and 2,3-disubstituted quinazolinones [22], Liu et al.,and Ioannis et al.,reported a microwave assisted synthesis of 2,3-disubstituted quinazolinones [27, 28],Adib et al.,reported the K2CO3 catalyzed reaction of isatoic anhydride,benzyl halides and amines [32] (Scheme 1).

|
Download:
|
| Scheme. 1.General approaches for the Synthesis of 2,3-di substituted quinazolinones. | |
Several reports available for the synthesis of 2,3-disubstituted quinazolinones,however,some of these multistep procedures have significant drawbacks such as long reaction times,low yield, harsh reaction conditions and use of expensive reagents [23- 26]. Development of simple and efficient methods for the synthesis of 2,3-disubstituted quinazolinones are more desirable. T3P was initially employed as peptide coupling agent and water scavenger with low toxicity and less allergic [33]. Its utility has been successfully demonstrated in rearrangement reactions [2], heterocyclic synthesis [2] and its shows versatile catalytic property [2]. In continuation of our work on synthetic applications of T3P [2],here in,we report the synthesis of various disubstituted 3H-quinazolin-4-ones which involving an in situ coupling of amines with anthranilic acid followed by the cyclocondensation of anthranilamide intermediate with different aldehydes,and further dehydrogenation gives 2,3-disubstituted quinazolinones.
2. Experimental
All of the reagents were used directly as obtained commercially. Column chromatography was performed with silica gel (100-200mesh) and analytical TLC on silica 60-F24. 1H NMR and 13C NMR spectra were determined in DMSO-d6 on a Bruker Fourier 300 MHz spectrometer (or Bruker Avance III 400 MHz) and chemical shifts (δ) reported relative to internal TMS.
General procedure: To a solution of anthranilic acid (1 mmol) in ethyl acetate (3 mL) solvent,triethyl amine (1.5 mmol) and T3P (2 mmol) was added and the resulting reaction mixture was stirred at room temperature for 1-2 h. Then,aldehyde (1 mmol) was added and stirred for another 1-2 h. The reaction was monitored by TLC. 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (1 mmol) was added to the reaction mixture and further stirred for 30 min. After completion of the reaction,the mixture is diluted with (20 mL) water and neutralized with 10% NaHCO3 solution. The product was extracted with ethyl acetate (10 mL) and the combined organic phase was washed with water (10 mL) and brine solution. The organic phase was dried over anhydrous Na2SO4. The solvent was dried under reduced pressure to afford a crude product,which was purified by column chromatography on silica gel using petroleum ether/ethyl acetate as eluent to provide the desired product. 2,3-Disubstituted 3H-quinazolin-4-ones 4a-4p are characterized by 1H NMR,13C NMR,elemental analysis and LCMS,the detail please see Supporting information.
3. Results and discussion
Initial investigations involved to determine the scope and optimal conditions for T3P/DDQ catalyzed synthesis of 2,3- disubstituted quinazolinones. Anthranilic acid 1a was selected as a model (Table 1),the reaction of 1a (1.1 equiv.),2a (1.0 equiv.), Et3N (1.5 equiv.) and T3P (2.0 equiv.,50% solution in EtOAc) in 10 volume of EtOAc at 25 °C for 2 h yielded an amide intermediate, which was then treated with an aldehyde 3d and DDQ were stirred for additional 2 h to gave a dihydro 2,3-disubstituted quinazolinone. Remarkably,the reaction proceeded efficiently even at room temperature. In fact,high yield of 4d was observed in this instance (Table 1,entry 4). A reduction in the loading of T3P to 1.5 equiv. showed negative effect on the yield of 4d (Table 1,entry 2), whereas increasing to 2.5 equiv. did not offer any significant advantage over the 2 equiv. catalyst loading (entry 3). Further screening of appropriate oxidants indicated that DDQ is superior to iodine,iron chloride (FeCl3),tert-butyl hydroperoxide (TBHP) were less effective in this oxidative dehydrogenation reaction. In the absence of DDQ,2,3-dihydroquinazolinone-4(1H)-one was formed and the results are summarized in Table 1.
| Table 1 Optimization of conditions for T3P catalyzed one pot three-component synthesis of 2,3-disubstituted 3H-quinazolin-4-ones.a |
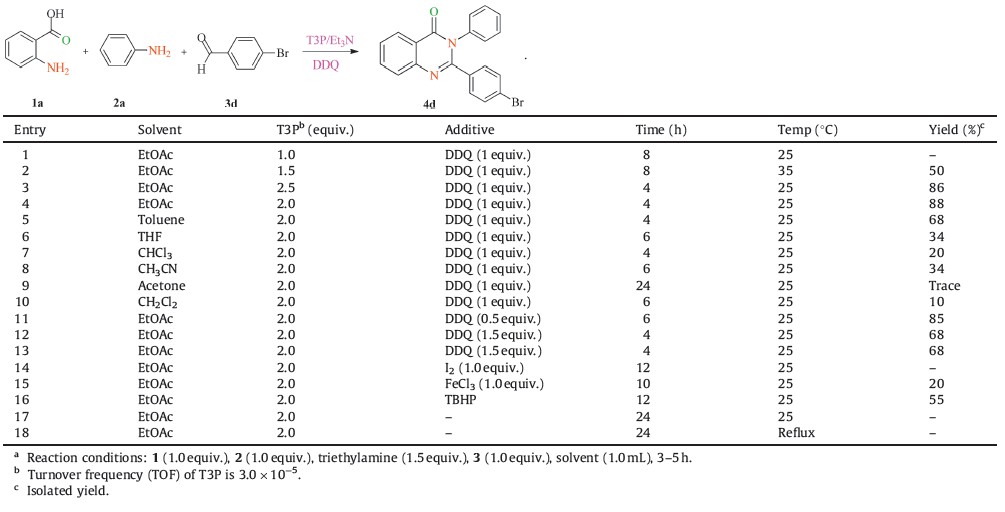
The influence of various solvents on the synthesis of 2,3- disubstituted quinazolinone 4d was studied in which ethyl acetate was chosen as the appropriate solvent with consideration of yield and the results are summarized in Table 1 (entry 4-10).
Under the established conditions,we evaluated the reactions of anthranilic acids (1a-c) ,amine (2a-f) ,and aldehydes (3a-g) . In all cases,products were obtained in good yield at room temperature. Finally,benzylic amines,allylamines,amino esters also underwent the title reaction under equally mild conditions. Furthermore,we found that the reaction showed good tolerance to the electronic properties of the substituents on the benzene ring of aldehydes. The target quinzolinones 4(a-p) were all formed in excellent yields,regardless of whether the aldehyde containing electronwithdrawing or electron-donating substituent’s (Table 2).
| Table 2 T3P catalyzed synthesis of 2,3-disubstituted quinazolinones. |

{kind=link}
{kind=link}
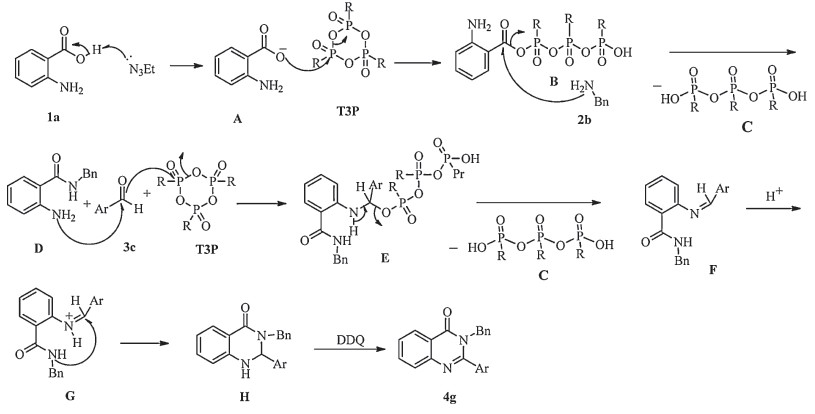
A possible mechanism of the coupling cyclization to get disubstituted quinazolinones suggested in Scheme 2. The catalytic reaction involves the activation of anthranilic acid 1a by T3P followed by reaction with amine to give anthranilamide. The second step in the probable mechanism of T3P catalyzed condensation of anthranilamide with aldehyde to afford an intermediate E,which generates an imine intermediate F. The byproduct P,P',P''-tripropyl triphosphonic acid C protonates imine to give protonated imine G. The next step involves intramolecualr cycloaddition to yield the key intermediate H,and the subsequent dehydrogenation to give the final 2,3-dihydroquinazolin-4(1H)- one 4g.

|
Download:
|
| Scheme. 2.Possible mechanism for T3P catalyzed synthesis of di-substituted quinazolinones. | |
{kind=link}
In summary,we developed a T3P catalyzed novel and straightforward methodology for the synthesis of 2,3-disubstituted quinazolinones by one-pot and three component reaction using anthranilic acid. This method features short reaction time,broad functional group tolerance,easy isolation,high yield and simple procedure.
AcknowledgmentFinancial support from DST-Fast track,New Delhi (No. SERB/F/ 2013-14) is gratefully acknowledged.
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.03. 037.
| [1] | A. Dö mling, Recent developments in isocyanide based multicomponent reactions in applied chemistry, Chem. Rev. 106 (2006) 17-89. |
| [2] | C. Hulme, V. Gore, Multi-component reactions: emerging chemistry in drug discovery ‘From Xylocain to Crixivan’, Curr. Med. Chem. 10 (2003) 51-80. |
| [3] | J. Zhu, Recent developments in the isonitrile-based multicomponent synthesis of heterocycles, Eur. J. Org. Chem. (2003) 1133-1144. |
| [4] | A. Dö mling, I. Ugi, Multicomponent reactions with isocyanides, Angew. Chem. Int. Ed. 39 (2000) 3168-3210. |
| [5] | (a) L. Weber, High-diversity combinatorial libraries, Curr. Opin. Chem. Biol. 4 (2000) 295-302; (b) A. Domling, Recent advances in isocyanide-based multicomponent chemistry, Curr. Opin. Chem. Biol. 6 (2002) 306-313. |
| [6] | S.B. Mhaske, N.P. Argade, The chemistry of recently isolated naturally occurring quinazolinone alkaloids, Tetrahedron 62 (2006) 9787-9826. |
| [7] | T. Onaka, A general three-step synthesis of pyrrolidino[2,1-b]quinazolone alkaloids via biogenetically patterned path, Tetrahedron Lett. 12 (1971) 4387-4390. |
| [8] | A. Hamid, A. Elomri, A. Daich, Expedious and practical synthesis of the bioactive alkaloidsrutaecarpine, euxylophoricine A, deoxyvasicinone and their heterocyclic homologues, Tetrahedron Lett. 47 (2006) 1777-1781. |
| [9] | J.R. Sheu, Pharmacological effects of rutaecarpine, an alkaloid isolated from evodia rutaecarpa, Cardiovasc. Drug Rev. 17 (1999) 237-245. |
| [10] | J. Michel, Quinoline, quinazoline and acridone alkaloids, Nat. Prod. Rep. 21 (2004) 650-668. |
| [11] | A.M. Al-Obaid, S.G. Abdel-Hamide, H.A. El-Kashef, et al., Substituted quinazolines, part 3. Synthesis, in vitro antitumor activity and molecular modeling study of certain 2-thieno-4(3H)-quinazolinone analogs, Eur. J. Med. Chem. 44 (2009) 2379-2391. |
| [12] | A.S. El-Azab, M.A. Al-Omar, A.A. Abdel-Aziz, et al., Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: molecular docking study, Eur. J. Med. Chem. 45 (2010) 4188-4198. |
| [13] | J.F. Wolfe, T.L. Rathman, M.C. Sleevi, J.A. Campbell, T.D. Greenwood, Synthesis and anticonvulsant activity of some new 2-substituted 3-aryl-4(3H)-quinazolinones, J. Med. Chem. 33 (1999) 161-166. |
| [14] | K. Terashima, H. Shimamura, A. Kawase, et al., Studies on antiulcer agents. IV: Antiulcer effects of 2-benzylthio-5,6,7,8-tetrahydro-4(3H)-quinazolinones and related compounds, Chem. Pharm. Bull. 43 (1995) 2021-2023. |
| [15] | J.B. Koepfli, J.F. Mead, J.A. Brockman Jr., An alkaloid with high antimalarial activity from dichroa-febrifuga, J. Am. Chem. Soc. 69 (1947) 1837. |
| [16] | S. Kobayashi, M. Ueno, R. Suzuki, H. Ishitani, Catalytic asymmetric synthesis of febrifugine and isofebrifugine, Tetrahedron Lett. 40 (1999) 2175-2178. |
| [17] | S.L. Cao, Y.P. Feng, Y.Y. Jiang, et al., Synthesis and in vitro antitumor activity of 4(3H)-quinazolinone derivatives with dithiocarbamate side chains, Bioorg. Med. Chem. Lett. 15 (2005) 1915-1917. |
| [18] | M.A.G. Nagwa, H.G. Hanan, M.Y. Riham, A.E.S. Nehad, Synthesis and antitumor activity of some 2,3-disubstituted quinazolin-4(3H)-ones and 4,6-disubstituted-1,2,3,4-tetrahydroquinazolin-2H-ones, Eur. J. Med. Chem. 45 (2010) 6058-6067. |
| [19] | C. Huang, Y. Fu, H. Fu, Y. Jiang, Y. Zhao, Highly efficient copper-catalyzed cascade synthesis of quinazoline and quinazolinone derivatives, Chem. Commun. 46 (2008) 6333-6335. |
| [20] | J.F. Liu, J. Lee, M.A. Dalton, et al., Microwave-assisted one-pot synthesis of 2,3-disubstituted 3H-quinazolin-4-ones, Tetrahedron Lett. 46 (2005) 1241-1244. |
| [21] | I.K. Kostakis, A. Elomri, E. Segunin, M. Iannelli, T. Besson, Rapid synthesis of 2,3-disubstituted quinazolin-4-ones enhanced by microwave-assisted decomposition of formamide, Tetrahedron Lett. 48 (2007) 6609-6613. |
| [22] | M. Adib, E. Sheikhi, H.R. Bijanzadeh, Benzyl halides, that are first oxidized to aldehydes under mild Kornblum conditions, undergo a three-component reaction with isatoic anhydride and primary amines to produce 4(3H)-quinazolinones in excellent yields, Synlett 23 (2012) 85-88. |
| [23] | A. Kumar, A.K. Bishnoi, Nanoparticle mediated organic synthesis (NAMO-synthesis): CuI-NP catalyzed ligand free amidation of aryl halides, RSC Adv. 4 (2014) 41631-41635. |
| [24] | H. Wei, T. Li, Y. Zhou, L. Zhou, Q. Zeng, Copper-catalyzed domino synthesis of quinazolin-4(3H)-ones from (hetero) arylmethyl halides, bromoacetate, and cinnamyl bromide, Synthesis 45 (2013) 3349-3354. |
| [25] | J. Zhou, L. Fu, M. Lv, et al., Copper(I) iodide catalyzed domino process to quinazolin-4(3H)-ones, Synthesis 24 (2008) 3974-3980. |
| [26] | J. Raid, J.V. Wolfgang, S. Muhammad, A novel method for the synthesis of 4(3H)-quinazolinones, Tetrahedron Lett. 45 (2004) 3475-3476. |
| [27] | H. Wissmann, H.J. Kleiner, New peptide synthesis, Angew. Chem. 92 (1980) 133-134. |
| [28] | R. Escher, P. Bunning, Synthesis of N-(1-Carboxy-5-aminopentyl)dipeptides as inhibitors of angiotensin converting enzyme, Angew. Chem. Int. Ed. 25 (1986) 277-278. |
| [29] | N. Basavaprabhu, R.S. Narendra, V.V. Lamani, Sureshbabu, T3P® (propylphosphonic anhydride) mediated conversion of carboxylic acids into acid azides and one-pot synthesis of ureidopeptides, Tetrahedron Lett. 51 (2010) 3002-3005. |
| [30] | B.S. Patil, G.R. Vasanthakumar, V.V. Sureshbabu, Isocyanates of N α-[(9-fluorenylmethyl) oxy]carbonyl amino acids: synthesis, isolation, characterization, and application to the efficient synthesis of urea peptidomimetics, J. Org. Chem. 68 (2003) 7274-7280. |
| [31] | J.K. Augustine, A. Bombrun, A.B. Mandal, et al., Propylphosphonic anhydride (T3P1)-mediated one-pot rearrangement of carboxylic acids to carbamates, Synthesis 9 (2011) 1477-1483. |
| [32] | M. Desroses, T. Koolmeister, S. Jacques, et al., A facile and efficient synthesis of tetrahydro-β-carbolines, Tetrahedron Lett. 54 (2013) 3554-3557. |
| [33] | T.M. Basavaprabhu, N.R. Vishwanatha, V.V. Panguluri, Sureshbabu, Propanephosphonic acid anhydride (T3P®) -a benign reagent for diverse applications inclusive of large-scale synthesis, Synthesis 45 (2013) 1569-1601. |
| [34] | G.M. Raghavendra, A.B. Ramesha, C.N. Revanna, et al., One-pot tandem approach for the synthesis of benzimidazoles and benzothiazoles from alcohols, Tetrahedron Lett. 52 (2011) 5571-5574. |
| [35] | A.B. Ramesha, G.M. Raghavendra, K.N. Nandeesh, K.S. Rangappa, K. Mantelingu, Tandem approach for the synthesis of imidazo[1,2-a]pyridines from alcohols, Tetrahedron Lett. 54 (2013) 95-100. |
| [36] | C.N. Revanna, G.M. Raghavendra, T.A. Jenifer Vijay, et al., Propylphosphonic anhydride-catalyzed tandem approach for biginelli reaction starting from alcohols, Chem. Lett. 43 (2014) 178-180. |