




Glycopolymers are synthetic polymers featuring pendant carbohydrate moieties on a non-carbohydrate main chain [1]. Their good water solubility,low toxicity and biocompatibility render them a wide range of applications,such as drug delivery [2], vaccines [3],fluorescent probes [4],etc. To further extend these bio-applications to a much boarder area,one feasible method is to hybridize them with inorganic nanomaterials [5, 6]. The corresponding "sweet" hybrid nanomaterials have shown great promise in the areas of sensor [7],magnetic resonance imaging [8] and vaccines [3],which made them receive quite a few attentions.
However,in most cases,thiol-functionalized glycopolymers were directly bound to gold nanoparticles (AuNPs) via Au-S bond. When surface of AuNP is covered with thiol-containing polymers, the nanoparticle only provides a scaffold and the property of AuNP itself could not be fully incorporated [9]. One convenient way to solve this problem is to stabilize AuNPs without using thiol group. In literature,monosaccharide on the surface of dendrimers was proved to stabilize AuNPs in water [10]. Recently,we also have demonstrated that AuNPs can be generated in situ and stabilized by glyco-inside vesicles [11]. This study provided a new route to generate rather "free" AuNPs within self-assembled micelles or vesicles,which might be beneficial for their further bio-applications. But in these studies,full manipulation on morphology of the hybrid nanomaterial was not achieved,which might hamper our further exploration. Thus,this paper aims at generating nanomaterials stabilized by glycopolymers with precise control on the morphology. By controlling the thickness of glyco-shell,Janus "sweet" nanoparticle was realized,which has not been reported in literature. In the end,catalysis property of these nanomaterials was also investigated. 2. Results and discussion 2.1. Synthesis and characterization of triblock glycopolymers
To control the morphology of hybrid nanomaterials [12] and even make Janus ones,polymer scientists have developed some intelligent strategies [13, 14, 15]. Among them,properties of the polymeric micelles,including the lengths of different stabilizing blocks and softness of stabilizing micelle cores were also employed [16]. However,glyco-NPs have not been utilized for preparation of Janus hybrid particles. Considering the bulky sugar pendant groups on the main chain of glycopolymers,we suppose that the effect from glyco-block might be much more significant than the previous non-sugar units. Thus two tri-block copolymers are designed,i.e. their middle blocks are glyco-blocks with hydrophobic and hydrophilic blocks on their both sides. By controlling the ratio between the sugar block and the hydrophobic block,different morphologies of hybrid nanomaterials (glyco-AuNPs) were expected after in situ reduction of precursor HAuCl4.
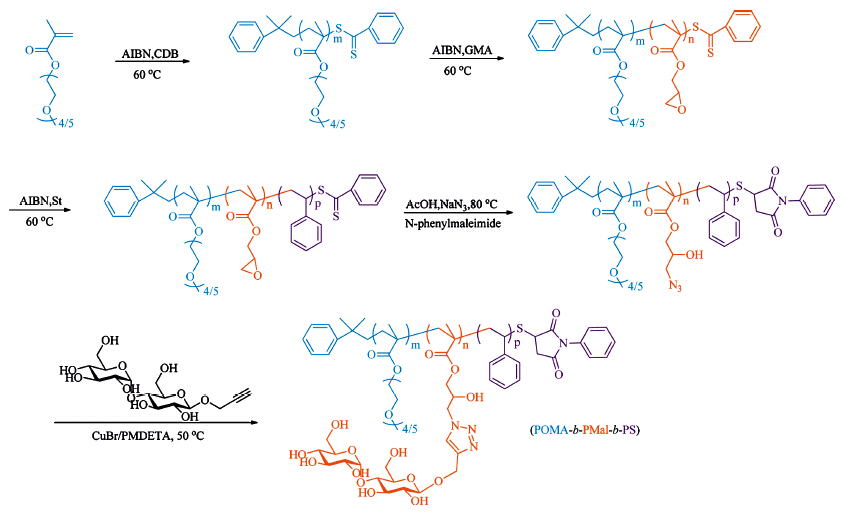
Glycopolymers were synthesized via the combination of reversible addition-fragmentation chain transfer (RAFT) polymerization and post-polymerization modification. As shown in Scheme 1,the triblock glycopolymer contains three components, from left to right,a hydrophilic poly(oligo(ethyleneglycol) methacrylate) (POMA) block,a hydrophilic glyco-block (PMal) and a hydrophobic polystyrene block (PS). The length of POMA in the two series of triblock glycopolymers was similar. However,the ratio of PMal:PS was designed to vary,which might help us to demonstrate the contribution of the middle glyco-block.

|
Download:
|
| Scheme 1.Synthetic route of amphiphilic triblock glycopolymer POMA-b-PMal-b-PS. | |
Briefly,oligo(ethylene glycol) methacrylate (OMA,Mn = 300 g mol-1) was polymerized by RAFT polymerization using cumyl dithiobenzoate (CDB) as chain transfer agent (CTA),yielding P1-1 and P2-1. As shown in Table 1,the degree of polymerization (DP) of POMA was obtained by comparison of the integral areas of ethylene glycol peak at 4.02 ppm and that of the phenyl group of CTA at 7.0-8.0 ppm in 1H NMR. Then,the obtained POMA was used as macro chain transfer agent (macro-CTA) to prepare the second block poly(glycidyl methacrylate) (PG),yielding two diblock copolymers P1-2 and P2-2. Similarly,the DP of PG block was calculated by comparing the 1H NMR peak of epoxy unit of PG at 2.64 ppm and that of ethylene glycol at 4.02 ppm. The triblock copolymer POMA-b-PG-b-PS was further prepared using the previous diblock copolymer (P1-2 and P2-2) as macro-CTA and styrene as monomer,yielding P1-3 and P2-3,with their DP obtained from the integral area of PS at 6.5-7.5 ppm. All of these copolymers showed narrow polydispersity indexes (PDI),measured by gel permeation chromatography (GPC). And the characterization results are shown in Table 1 and Figs. S1 and S2 in Supporting information. It is worth to mention that 1H NMR results were used to determine DP,since PEG was used as calibration standard for GPC analysis,which was not effective to characterize glycopolymers due to great structural inconsistency.
| Table 1 Mn and PDI of precursors of the final triblock copolymers. |

{kind=link}
Combination of ring-opening reaction and click reaction was used in post-polymerization modification. P1-3 and P2-3 were treated with NaN3 and acetic acid at 80℃ to obtain azidecontaining copolymer P1-4 or P2-4,by the ring opening of epoxide from the glycidyl methacrylate. Disappearance of the epoxy units in 1H NMR spectra and the appearance of azide at 2105 cm-1 in Fourier transform infrared spectra (FT-IR) demonstrated the complete transformation from epoxide to azide (Fig. S3 in Supporting information). After click reaction with propargyloxyb- β-maltopyranoside,the strong peak of azide at 2105 cm-1 disappeared (Fig. S3),while the peak at 3340 cm-1 increased due to the hydroxyl groups from maltose. This result indicated the complete conversion to POMA-b-PMal-b-PS. 1H NMR was also used to characterize the triblock glycopolymers (Fig. S3). Finally,a pair of triblock glycopolymers was determined as POMA22-b-PMal54-b- PS17 (P1) with PMal to PS ratio as 3:1,and POMA20-b-PMal26-b- PS108 (P2) with PMal to PS ratio as 1:4. As control,POMA22-b- PPA54-b-PS17 (P3) was obtained by the same strategy but propargyl alcohol was used to click with azide-containing polymer P1-4 (FTIR and 1H NMR in Fig. S4 in Supporting information) instead. What is more,diblock copolymer PMal47-b-PS158 (P4) was also synthesized (GPC,FT-IR and 1H NMR characterization data are shown in Fig. S5 and S6) via the similar strategy and used as another control. 2.2. Preparation of glyco-NPs
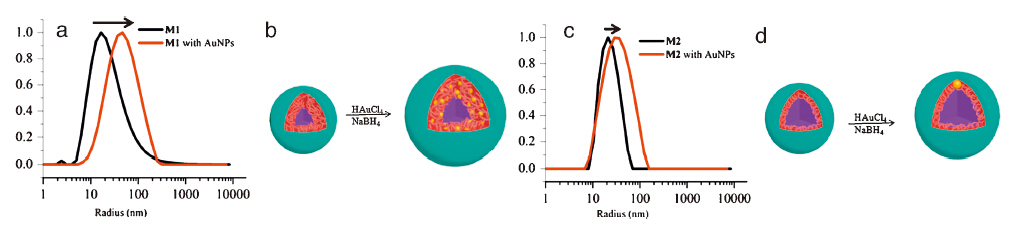
Self-assembly of glycopolymers were performed by selective
solvent method. Typically P1 or P2 was firstly dissolved in 1 mL
DMAc (dimethylacetamide,10 mg mL-1),then 8 mL deionized
water was added into the solution quickly. After dialysis,the final
concentration was fixed at 0.83 mg mL-1 in which self-assembled structures were obtained. For clarity,M1 and M2 were used to
represent the nanostructures formed by the corresponding
glycopolymer precursors P1 and P2,respectively. Dynamic light
scattering (DLS) and transmission electron microscopy (TEM) were
used to characterize these structures (Fig. 1 and Fig. S7 in
Supporting information). DLS measurements showed that
M1 and M2 were used for in situ AuNP formation. Chloroauric
acid (HAuCl4) was firstly mixed with the glyco-NPs by stirring at
room temperature overnight. UV-vis spectrum of the mixture
proved that no self-reduction happened during this process (Fig.
S10 in Supporting information). Then NaBH4 (3.2 equiv. to HAuCl4)
was then added and the mixture turned brown in several seconds.
Appearance of a distinct characteristic absorption peak at 515 nm
in the UV-vis spectrum (Fig. S11 in Supporting information)
confirmed the formation of AuNPs. TEM images also demonstrated
the presence of AuNPs,i.e. the black dots with diameters smaller
than 10 nm (Fig. 2). The magnified images of the AuNPs showed an
obvious crystal lattice and the inter-planar spacing of the lattice
was near 0.224 nm (Fig. 2b and d),which agreed well with the
(1 1 1) lattice spacing of AuNPs. The EDX (energy dispersive X-ray)
spectrum showed that there were strong peaks for elemental gold
indicating that these nanoparticles were composed of Au (Fig. S12
in Supporting information). It was quite interesting to find that M1
with a thick sugar shell had many AuNPs with diameter around
3 nm(Fig. 2a and b with inset cartoon). While most of M2 with thin
sugar shells had only one AuNP with diameter around 7 nm(Fig. 2c
and d with inset cartoon),demonstrating the feature of Janus
particles. This "one-micelle-one-AuNP" hybrid structure could be
easily found under TEM (Fig. 2c and d) and more TEM images were
presented in Fig. S13 in Supporting information. Interestingly,after
reduction of HAuCl4,the To investigate the role of glyco-shell thickness in nanoparticle
hybridization,different amounts of HAuCl4 were employed to form
glyco-AuNPs,while the amount of the glyco-NPs and reducing
reagent NaBH4 were constant. To the aqueous solution of glyco-
NPs (1 mL,0.83 mg mL-1),different amount of HAuCl4
(50 mg mL-1) was added (1 μL,5 μL and 10 μL),followed by
incubation and reduction. As shown in Fig. S16 in Supporting
information,the size of AuNPs encapsulated by M1 was all around
3 nm (Fig. S16(a)-(c)),and more AuNPs were found inside the
micelles with the increasing concentration of HAuCl4. On the
contrary,the encapsulated amount of AuNPs by M2 only slightly
increased with increasing concentration of HAuCl4 (Fig. S16(d)-
(f)). The result indicated that the Janus particle morphology was
greatly related to the thickness of glyco-shell,not directly
determined by the concentration of HAuCl4 and NaBH4. At an
appropriate concentration of HAuCl4 and NaBH4,Janus particles
would be observed in the presence of M2 (Fig. 2c and d). For the mechanism,we supposed that HAuCl4 might locate in the sugar
layer and protected by the POMA corona. After reduction,if the
sugar layer was very thin,only a few Au seeds could be loaded in
the shell and only one seed can grow up forming nanoparticle.
However,when the sugar shell became thicker,more Au seeds
would be inside the shell resulting in formation of much more
AuNPs.
2.4. Catalytic reduction of 4-nitrophenol (4-NP)
To evaluate the catalytic activity of the glyco-AuNPs,model
reduction reaction of 4-nitrophenol (4-NP) to 4-aminophenol (4-
AP) in the presence of an excess amount of NaBH4 was studied.
AuNPs were widely used as catalyst for this reaction (Fig. S17 in
Supporting information) by relaying electrons from donor BH4
-to acceptor 4-NP [17],when both of them were adsorbed on surface.
The reaction process was monitored by UV-vis spectrometry. As
shown in Fig. S18c in Supporting information,the reaction could
not happen in the absence of glyco-AuNPs. After addition of the
catalyst,the characteristic peak of 4-NP at 400 nm started to
decrease,while a new peak of 4-AP at 295 nm appeared (Fig. S18).
After a while,the peak of 4-NP was no longer observed and the
solution color changed from yellow to colorless,indicating that the
catalytic reduction of 4-NP proceeded completely. The rate
constant k was calculated using the rate law for the first-order
kinetics: ln(At/A0) = -kt,where At represented absorbance of 4-NP
at any time,A0 represented the starting absorbance of 4-NP and
k was rate constant. Fig. 3 showed a good linear correlation of
ln(At/A0) versus time,therefore the kinetic reaction rate constant
was estimated as 4.0 × 10-3 s-1 for M1 and 2.2 × 10-3 s-1 for
M2. Both the them were larger than citrate-capped AuNPs
(1.2 ± 0.1 × 10-3 s-1) with diameter of 3.5 nm and poly(N-isopropylacrylamide)-
b-poly(4-vinyl pyridine) (PNIPAM-b-P4VP) stabilized
AuNPs (1.5 × 10-3 s-1) with diameter of 3.3 nm,which located in the
dense core of P4VP [18, 19].
3. Conclusion
A novel amphiphilic brush triblock glycopolymer POMA-b-
PMal-b-PS was prepared through the combination of RAFT
polymerization and post-polymerization modification. Micelles
prepared by this new glycopolymer were used to in situ formation
of AuNPs. Different morphologies including Janus particles and
raspberry-like particles were obtained by changing the ratio of
PMal and PS.
Acknowledgments
Ministry of Science and Technology of China (No.
2011CB932503332),National Natural Science Foundation of China
(No. 91227203,21474020 and 51322306),and the Shanghai
Rising-Star Program (No. 13QA1400600) are acknowledged for
their financial support.
Appendix A. Supplementary data
Supplementary data associated with this article can be found,in
the online version,at http://dx.doi.org/10.1016/j.cclet.2015.05.022.

Fig. 1.(a) DLS and (b) cartoon scheme of M1 before and after reduction of HAuCl4; (c) DLS and (b) cartoon scheme of M2 before and after reduction of HAuCl4.

Fig. 2.TEM images of different glyco-AuNPs. (a), (b) M1 with many AuNPs inside (inset: cartoon scheme); (c), (d) M2 with only one AuNP inside (inset: cartoon scheme).

Fig. 3.Plot of ln(At/A0) versus time for the reduction of 4-NP. Black line: M1 with AuNPs; Red line: M2 with AuNPs.
{kind=link}
{kind=link}
{kind=link}
| [1] | A. Ghadban, L. Albertin, Synthesis of glycopolymer architectures by reversibledeactivation radical polymerization, Polymers 5 (2013) 431–526. |
| [2] | C. Zheng, Q.Q. Guo, Z.M. Wu, et al., Amphiphilic glycopolymer nanoparticles as vehicles for nasal delivery of peptides and proteins, Eur. J. Pharm. Sci. 49 (2013) 474–482. |
| [3] | A.L. Parry, N.A. Clemson, J. Ellis, et al., 'Multicopy Multivalent' glycopolymerstabilized gold nanoparticles as potential synthetic cancer vaccines, J. Am. Chem. Soc. 135 (2013) 9362–9365. |
| [4] | T. Xing, X.Z. Yang, L.Y. Fu, L.F. Yan, Near infrared fluorescence probe and galactose conjugated amphiphilic copolymer for bioimaging of HepG2 cells and endocytosis, Polym. Chem. 4 (2013) 4442–4449. |
| [5] | M.H. Mashhadizadeh, R.P. Talemi, Application of diazo-thiourea and gold nanoparticles in the design of a highly sensitive and selective DNA biosensor, Chin. Chem. Lett. 26 (2015) 160–166. |
| [6] | X.H. Lv, L.P. Wang, G. Li, et al., Preparation and characterization of optically functional hollow sphere hybrid materials by surface-initiated RATRP and "click" chemistry, Chin. Chem. Lett. 24 (2013) 335–337. |
| [7] | M. Takara, M. Toyoshima, H. Seto, Y. Hoshino, Y. Miura, Polymer-modified gold nanoparticles via RAFT polymerization: a detailed study for a biosensing application, Polym. Chem. 5 (2014) 931–939. |
| [8] | A. Pfaff, A. Schallon, T.M. Ruhland, et al., Magnetic and fluorescent glycopolymer hybrid nanoparticles for intranuclear optical imaging, Biomacromolecules 12 (2011) 3805–3811. |
| [9] | K. Kuroda, T. Ishida, M. Haruta, Reduction of 4-nitrophenol to 4-aminophenol over Au nanoparticles deposited on PMMA, J. Mol. Catal. A: Chem. 298 (2009) 7–11. |
| [10] | K. Esumi, T. Hosoya, A. Suzuki, K. Torigoe, Spontaneous formation of gold nanoparticles in aqueous solution of sugar-persubstituted poly(amidoamine) dendrimers, Langmuir 16 (2000) 2978–2980. |
| [11] | L. Su, C.M. Wang, F. Polzer, et al., Glyco-inside micelles and vesicles directed by protection–deprotection chemistry, ACS Macro Lett. 3 (2014) 534–539. |
| [12] | Y.X. Zhang, X.D. Hao, Z.P. Diao, Templated self-assembly of Au–TiO2 binary nanoparticles–nanotubes, Chin. Chem. Lett. 25 (2014) 874–878. |
| [13] | T. Chen, M.X. Yang, X.J. Wang, L.H. Tan, H.Y. Chen, Controlled assembly of eccentrically encapsulated gold nanoparticles, J. Am. Chem. Soc. 130 (2008) 11858–11859. |
| [14] | J. He, M.T. Perez, P. Zhang, et al., A general approach to synthesize asymmetric hybrid nanoparticles by interfacial reactions, J. Am. Chem. Soc. 134 (2012) 3639– 3642. |
| [15] | W.P. Li, V. Shanmugam, C.C. Huang, et al., Eccentric inorganic-polymeric nanoparticles formation by thermal induced cross-linked esterification and conversion of eccentricity to raspberry-like Janus, Chem. Commun. 49 (2013) 1609–1611. |
| [16] | N. Ali, S.Y. Park, Micellar structures of poly(styrene-b-4-vinylpyridine)s in THF/ Toluene mixtures and their functionalization with gold, Langmuir 24 (2008) 9279–9285. |
| [17] | T. Premkumar, K. Lee, K.E. Geckeler, Shape-tailoring of gold nanostructures: can a detergent act as the reducing or protecting agent? Nanoscale 3 (2011) 1482– 1484. |
| [18] | R. Fenger, E. Fertitta, H. Kirmse, A.F. Thü nemann, K. Rademann, Size dependent catalysis with CTAB-stabilized gold nanoparticles, Phys. Chem. Chem. Phys. 14 (2012) 9343–9349. |
| [19] | Y.Wang, G.W.Wei, W.Q. Zhang, et al., Responsive catalysis of thermoresponsive micelle-supported gold nanoparticles, J. Mol. Catal. A: Chem. 266 (2007) 233–238. |