



 , Zhan-Ting Li
, Zhan-Ting Li
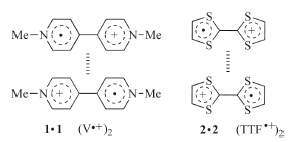
Stacking is a common phenomenon for conjugated molecules or segments. The driving force is usually electrostatic attraction between electron-rich aromatic donor(s) and electron-deficient aromatic acceptor(s) or solvophobicity,as observed in water for the base pairs of DNA and the aromatic units of proteins. One unconventional stacking pattern involves the homodimerization of conjugated radical cations,which has been demonstrated to be driven by the inherent multicenter covalent π-π bonding of the radical species or the electrostatic attraction of paired anions in the solid state [1]. In 1964,Kosower and co-workers reported the dimerization of viologen (V2+) (1) radical cation [2],and in 1979, Bozio and co-workers described the dimerization of tetrathiafulvalene (TTF) (2) radical cation [3] (Fig. 1). However,the stability of both prototypical radical cation dimers was low and thus they had been observed only in the solid state or in concentrated solutions at low temperatures. In the past decade,several strategies have been developed to enhance this self-binding motif of the V·+ and TTF·+ radical cations,which include using a rigid macrocycle or capsule to encapsulate the dimer [4],holding two radical cation units in place with a preorganized framework [5],and stabilizing the dimer utilizing mechanical bonding in interlocked systems [6]. In addition to the enhanced stability in both organic and aqueous media,the dimerization of the V·+ and TTF·+ radical cations can be conveniently monitored by recording the absorption of the dimers in the visible light area. As a result,this unique non-covalent interaction has been developed as a new useful driving force for the construction of supramolecular architectures. This short review summarizes the recent advance in this field.

|
Download:
|
| Fig. 1.The structure of dimer of viologen (1) and TTF (2) radical cation. | |
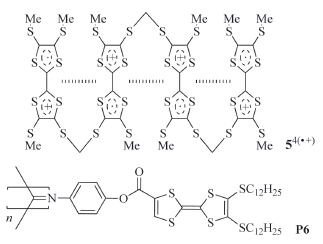
Solvophobically driven aromatic stacking usually occurs in polar or less polar solvents,but is weak in the so-called "good" solvents,such as chloroform. CRCD can take place in nearly all solvents of low and high polarity and thus provides a new approach to designing supramolecular systems. Becher and co-workers reported that TTF·+ units incorporated in a macrocycle could undergo strong intramolecular stacking in acetonitrile [7]. Hasegawa and co-workers also found that the two TTF·+ units in naphthalene-based 32(·+) (Fig. 2) was forced to arrange in a face-toface style and formed a stable intramolecular dimer in a 1:1 mixture of dichloromethane and acetonitrile,which was supported by the appearance of a typical absorption around 732 nm in the absorption spectrum [8]. We recently found that intramolecular hydrogen bonding-induced preorganization of 42(·+) (Fig. 2) could drive the two appended TTF·+ units to stack intramolecularly into (TTF·+)2 in less polar solvents like dichloromethane [9, 10, 11]. Successive stacking of the TTF·+ subunits can take place for linear molecules that contain multiple TTF subunits. For example, Misaki and co-workers showed that the two inner TTF·+ subunits of compound 54(·+) (Fig. 3) could stack from both sides in benzonitrile [12]. Amabilino and coworkers demonstrated that,upon oneelectron oxidation of the TTF subunits,the resulting TTF·+ subunits of polymer P6 (Fig. 3) also underwent intramolecular stacking in benzonitrile [13].

|
Download:
|
| Fig. 2.The structure of radical cations 32(·+) and 42(·+), highlighting the intramolecular stacking of the TTF·+ units. | |

|
Download:
|
| Fig. 3.The structure of radical cation 54(·+) and P6. | |
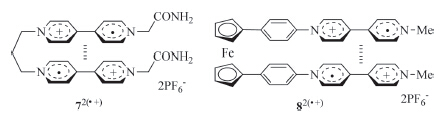
Such intramolecular stacking has also been observed for viologen derivatives. For example,Bucher and co-workers prepared viologen radical cation derivative 72(·+) (Fig. 4) [14] and found that in DMF or DMSO,the two V·+ subunits underwent intramolecular stacking to produce a folded conformation. Bucher et al. also reported that ferrocene-derived biradical cation 82(·+) (Fig. 4) formed the similar intramolecular stacking of the V·+ subunits [15]. Upon oxidation of the V·+ subunits to V2+,the two V2+ subunits were repelled electrostatically to lead to an extended conformation. Thus,the compound gave rise to a redox-tuned reversible switching system. Winter and co-workers also reported that bisviologen radical cations connected with a tetramethylene linker formed intramolecular (V·+)2 dimer at room temperature and,when the temperature was increased to 95℃,the dimer decomposed to produce an extended conformation [16]. Wang and co-workers reported that V·+ subunits appended to the periphery of hyperbranched polyglycerols exhibited enhanced intramolecular dimerization [17],even though the flexibility of the dendrimeric framework restricted the establishment of an explicit stacking pattern. Trabolsi and co-workers prepared phosphazene derivative 96·+ (Fig. 5) by reducing the appended six viologen subunits to V·+ with sodium hydrosulfite [18]. It was demonstrated that the two V·+ subunits connected to the same P atom formed one pair of dimer in the intramolecular way. The three pairs of dimers were very stable and did not decompose in the presence of cucurbit[7]uril (CB[7]),which is a good host to encapsulate one V·+ subunit.

|
Download:
|
| Fig. 4.The structure of radical cations 72(·+) and 82(·+), highlighting the intramolecular stacking of the V·+ units. | |

|
Download:
|
| Fig. 5.The structure of radical cation 96(·+), having three V·+ dimers. | |
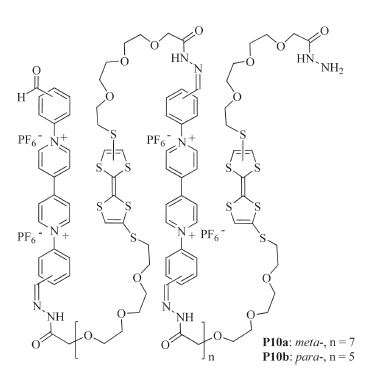
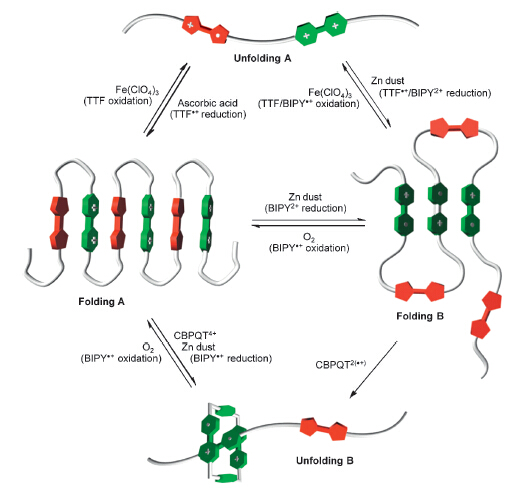
Such intramolecular radical cation stacking also occurs in long linear polymers. For example,Chen and co-workers prepared polymers P10a and P10b (Fig. 6) by forming hydrazine bonds from the corresponding di(acyl hydrazide) and dialdehyde precursors [19]. In acetonitrile,both polymers formed pleated foldamers due to intramolecular donor-acceptor interaction between the electron- rich TTF subunit and the electron-deficient V subunit (Folding A,Fig. 7). When the TTF subunits were oxidized to TTF·+,the polymers did not form confined conformations (Unfolding A, Fig. 7). In contrast,when the V2+ subunits were reduced to V·+, these V·+ subunits underwent intramolecular stacking to induce the backbones to form another kind of pleated conformations (Folding B,Fig. 7). Upon adding diradical dicationic cyclobis- (paraquat-p-phenylene) cyclophane (CBPQT2(·+)),these pleated foldamers were forced to unfold due to the inclusion of the V·+ subunits of the polymers by CBPQT2(·+) which was driven by the encapsulation-enhanced intermolecular stacking of the V·+ subunits [20]. The processors were reversible and confirmed by systematic UV/vis experiments.

|
Download:
|
| Fig. 6.The structure of polymers P10a and P10b. | |

|
Download:
|
| Fig. 7.Cartoons for multiply tuned folding-unfolding processes of polymers P10a and P10b driven by the intramolecular donor-acceptor interaction and TTF·+ or MV·+ dimerization. | |
Intermolecular stacking of simple radical cation species is usually very weak. Kochi et al. reported that in acetone the association constant (Ka) for the dimerization of TTF·+ was only 0.6 L/mol at 2℃ [21],reflecting the low stability of this prototypical dimer. By preorganizing the molecular framework and introducing more than one TTF subunits into one molecule to endow multivalence,stable dimeric complexes can be generated. Chiu and co-workers prepared rigid molecular clip 11 (Fig. 8) which bears two TTF subunits [22]. When the TTF subunits were oxidized to TTF·+,the molecule formed an interdigitated dimer stabilized by intermolecular multiple radical stacking interactions as confirmed by UV/vis and MS studies. However,no quantitative investigation was performed for this dimer.

|
Download:
|
| Fig. 8.The structure of clip-shaped dimer formed by compound 11. | |
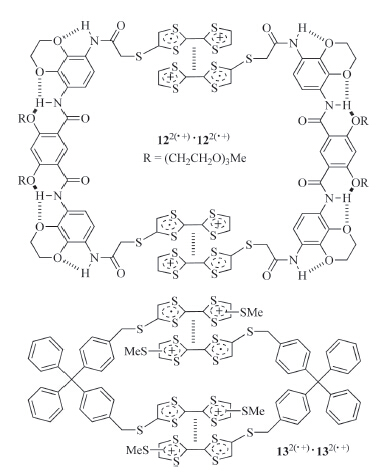
Extended monomers can also form dimeric aggregates if the linker connecting the radical cation subunits is preorganized. For example,Wang and co-workers prepared compound 12 (Fig. 9) by attaching two TTF subunits to a hydrogen bonding-induced preorganized aromatic amide linker [11]. After the TTF subunits underwent one-electron oxidation,the molecule could selectively form a bimolecular macrocycle (12·+)2 stabilized by the dimerization of the TTF·+ subunits in the mixture (1:1) of dichloromethane with acetone,ethanol or acetonitrile. The apparent Ka values for the TTF·+ dimerization were determined to be 1.0 × 104,3.9 × 103,and 5.9 × 102 L/mol,respectively. In contrast,simple TTF control did not exhibit detectable π-dimerization,indicating that the two TTF·+ subunits of 12 promoted each other for intermolecular dimerization. Tian and co-workers prepared another ditopic compound 13 (Fig. 9) [23]. When the TTF unit was oxidized to TTF·+,the two TTF·+ subunits did not dimerize intramolecularly due to the rigidity of the linker. Instead,it also formed a 1 + 1 macrocycle (13·+)2 through intermolecular dimerization of the TTF·+ subunits. In chloroform and acetonitrile,the apparent Ka of the TTF·+ dimerization was determined to be 6.3 × 104 and 4.5 × 103 L/mol,respectively. Intriguingly,no similar dimerization was observed in dichloromethane.

|
Download:
|
| Fig. 9.The structure of the dimeric macrocycles formed by radical cations 122(·+)and 132(·+). | |
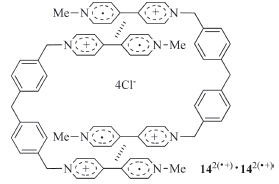
Zhou and co-workers extended the above approach for dimeric structures to viologen systems. They prepared water-soluble radical cation 142(·+) (Fig. 10) by reducing the corresponding bisviologen derivative [24]. In water,THF,1,4-dioxane,ethanol and methanol,142(·+) formed dimeric macrocycle [142(·+)]2 (Fig. 10), with the apparent Ka of V·+ dimerization being 7.6 × 106,9.5 × 104, 3.4 × 105,2.9 × 105,and 3.7 × 105 L/mol,respectively. In the above organic solvents,the radical cation of 1-methyl-4-benzyl-1,4- pyridinium did not dimerize. In water,the Ka for this dimerization was 1.0 × 103 L/mol,which is remarkably lower than that of 142(·+),reflecting the cooperativity of the V·+ units in dimerization.
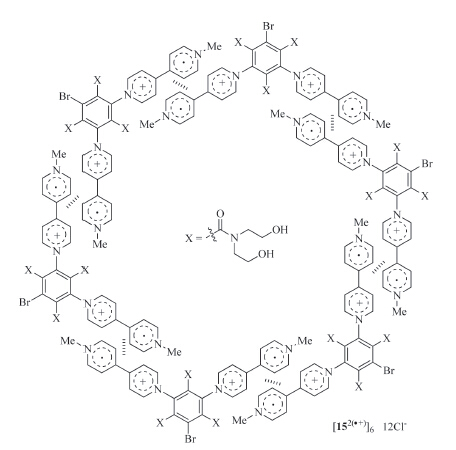
Zhang and co-workers reported that positive cooperation of the radical cation stacking was also exhibited by rigid V-shaped ditopic 152(·+) (Fig. 11) [25]. In water,the apparent Ka of its two V·+ subunits in dimerization was estimated to be 2.5 × 104 L/mol, which was also considerably higher than that of the radical cation of 1-methyl-4-phenyl-1,4-pyridinium (3.5 × 103 L/mol). Due to the existence of the three amide chains,152(·+) could not undergo face-to-face stacking. The cooperative stacking of its two V·+ subunits supported that it gave rise to the more complicated sixmembered macrocyclic complex (Fig. 11).

|
Download:
|
| Fig. 10.The structure of the dimeric macrocycle formed by radical cation 142(·+). | |

|
Download:
|
| Fig. 11.The structure of the hexameric macrocycle formed by radical cation 152(·+). | |
The TTF·+ or V·+ subunits of tritopic monomers 163(·+) and 173(·+) also underwent strong intramolecular dimerization (Fig. 12) [23, 24]. In all the solvents investigated,the apparent Ka corresponding to this dimerization was higher than that of the related ditopic 132(·+) and 142(·+). Thus,a 1 + 1 capsular binding pattern had been proposed for the complex formed by 163(·+) and 173(·+) which could be stabilized by three pairs of radical cation stacking.

|
Download:
|
| Fig. 12.The structure of tritopic radical cation monomers 163(·+) and 173(·+). | |
To investigate the stacking of multiple TTF·+ subunits in one molecular system,Zhang and co-workers prepared extended and planar compounds 18-21 and polymer P22 (Fig. 13) by forming hydrazone bonds from the corresponding acylhydrazine and aldehyde precursors [26]. The introduction of the long oligoglycol chains endowed solubility in organic solvents after the TTF units were oxidized to TTF·+. The (apparent) Ka’s for the dimerization of the TTF·+ subunits were determined in the binary solution of chloroform and acetonitrile of changing ratio (Table 1). The results show that,in pure chloroform,intermolecular TTF·+ dimerization occurred for all the molecules,even though the longer ones exhibited increased capacity. With the addition of acetonitrile,the dimerization generally weakened. When the content of acetonitrile was increased to 15%,only the TTF·+ subunits of polymer P22 still dimerized considerably. This observation clearly suggests that the dimerization of the TTF·+ subunits in the long oligomers or polymer possesses the character of multicovalence.

|
Download:
|
| Fig. 13.The structure of compounds 18-21 and polymer P22. | |
| Table 1 Apparent association constants (Ka) of the TTF·+ units in the mixture of acetonitrile and chloroform. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
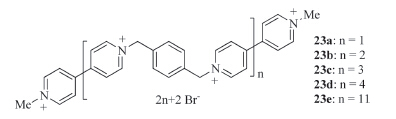
Stoddart and co-workers prepared viologen oligomers 23a-e (Fig. 14) and reduced the incorporated 4,4'-bipyridinium subunits to radical cations [27]. They found that the radical cations strongly stacked to induce intra- and intermolecular folding,which was confirmed by UV/vis/NIR spectroscopic studies. Interestingly,the folding of the shorter oligomers was dominated by intermolecular V·+ stacking,while strong intramolecular V·+ stacking,which gave rise to an NIR absorption band at 900 nm,tended to play a crucial role in governing the folding of the longer oligomers. The solidstate superstructure of 23b in the radical cation state revealed that two intertwining chains folded together to form a dimer,stabilized by intermolecular V·+ stacking.

|
Download:
|
| Fig. 14.The structure of viologen oligomers 23a-e. | |
{kind=link}
Multi-topic hydrogen bonding- or hydrophobicity-driven monomers are common in creating extended 2D or 3D supramolecular polymers. CRCD has also found applications in designing symmetric tritopic and tetratopic monomers. Compared to that of simple molecules,the interaction can be enhanced remarkably. In this context,Tian and co-workers prepared tetratopic derivatives 24a and 24b (Fig. 15) [23]. After the TTF subunits were oxidized to TTF·+,both molecules could self-assemble into 3D supramolecular polymers through the intermolecular dimerization of TTF·+. The apparent Ka 's for the TTF·+ dimerization were also evaluated in different solvents. The values were all larger than that of tritopic 163(·+) in the same solvent,reflecting enhanced cooperativity of the TTF·+ stacking of these tetratopic monomers. Dynamic light scattering (DLS) revealed that the two supramolecular polymers possessed a hydrodynamic diameter of 68 nm for 24a4(·+) (75 mmol/L) in acetonitrile and 105 nm for 24b4(·+) (75 mmol/L) in water and acetonitrile (1:1). Chen and co-workers further prepared 25a and 25b to investigate the effect of the linkers on the intermolecular stacking of the TTF·+ subunits [28],in which four TTF subunits were connected to the central tetraphenylmethane by the more rigid amide or ethynyl linker. After the TTF subunits were oxidized to TTF·+,they also self-assembled into 3D supramolecular polymers. The apparent Ka values of their TTF·+ dimerization in chloroform were also determined,which was lower than that of 24a,implying that a fully rigid framework might not be the best choice for maximizing the intermolecular TTF·+ stacking. Zhou and co-workers also prepared a similar tetrahedral monomer that bears four viologen subunits [24]. The viologen subunits could be reduced to radical cations in water. UV/vis and DLS experiments indicated that it also self-assembled into 3D supramolecular polymer driven by the intermolecular stacking of the radical cations.

|
Download:
|
| Fig. 15.The structure of compounds 24a,b and 25a,b. | |
{kind=link}
Recently supramolecular organic frameworks (SOFs) have been established as a newfamily of periodic self-assembled architectures in solution [29]. Zhang and co-workers prepared rigid triangular V·+ derivatives 26a3(·+) and 26b3(·+) (Fig. 16) [25]. In water,their V·+ subunits stacked strongly. For 26b3(·+),this stacking led to the formation of 1D supramolecular aggregates. For 26a3(·+),the three amide groups prevented similar 1D stacking and the molecules stacked in the 2D space to give rise to 2D supramolecular organic framework. The apparent Ka of this dimerization at 25℃ was determined to be 1.2 × 106 L/mol. The valuewas remarkably higher thanthat (3.5 × 103 L/mol) of amonomeric control,againsuggesting positive stacking cooperativity. CB[8] further enhanced the dimerization through encapsulation and thus stabilized the 2D SOF structure. The periodicity of the SOF was confirmed by UV-vis absorption,DLS,solution and solid phase small angle X-ray diffraction experiments.

|
Download:
|
| Fig. 16.The structure of planar and rigid tritopic radical cation monomers 26a3(·+)and 26b3(·+). | |
{kind=link}
In this short review,we have summarized the application of the dimerization of tetrathiafulvalene and viologen radical cations in constructing ordered unimolecular and supramolecular architectures. One feature of such CRCDis its orthonogality,which allows other non-covalent interactions to co-exist and to work separately or in a cooperativemanner. Thus,one promising aspect in the future research is the combination of CRCD with other interactions to produce more complicated and/or advanced structures. Another valuable feature of CRCD is that the dimers give rise to characteristic absorption band in the long-wavelength range,which makes its detection very convenient by using the absorption spectroscopy. For viologen radical cations,the dimerization may occur in aqueous solution. The feature is also important and expected to find more applications in the future because many binding motifs that work well in organic solvents become less stable or even decompose due to the competition of polar solvent molecules [30, 31, 32]. Although this paper focuses on CRCD of tetrathiafulvalene and viologen,the stacking of other conjugated radical cations,like oligothiophene radical cation,has also received attention in recent years. Given the unique features of CRCD,the development of new CRCD patterns should also be of high value in the future. The formation of conjugated radical cations concerns the oxidation or reduction of their aromatic ring precursors. The process is reversible,which also makes it an attractive choice to use CRCD to build tunable supramolecular systems.
Acknowledgments
We are grateful to the Ministry of Science and Technology of the China (No. 2013CB834501),the Science and Technology Commis-sion of Shanghai Municipality (No. 13M1400200),the Ministry of Education Research Fund for the Doctoral Program of China,and the National Natural Science Foundation of China (Nos. 21472023, 21432004) for financial support.
| [1] | (a) I. Garcia-Yoldi, J.S. Miller, J.J. Novoa, Theoretical study of the electronic structure of[tetrathiafulvalene]22+ dimers and their long, intradimer multicenter bonding in solution and the solid state, J. Phys. Chem. A 113 (2009) 484–492; (b) W. Lu, Q.Y. Zhu, J. Dai, et al., Tetrathiafulvalene-diamide salts with S…S and C…C stacked radical couples, Cryst. Growth Des. 7 (2007) 652–657; (c) F. Gao, F.F. Zhu, X.Y. Wang, et al., Stabilizing radical cation and dication of a tetrathiafulvalene derivative by a weakly coordinating anion, Inorg. Chem. 53 (2014) 5321–5327; (d) B.Q. Wang, F.F. Wang, F. Ma, et al., Intermolecular covalent interaction: 20- center-2-electron covalent p/p bonding in tetrathiafulvalene radical-cation dimer TTF·+–TTF·+, J. Comput. Methods Sci. Eng. 10 (2010) 357–366; (e) D.W. Zhang, J. Tian, L. Chen, L. Zhang, Z.T. Li, Dimerization of conjugated radical cations: an emerging non-covalent interaction for self-assembly, Chem. Asian J. 10 (2015) 56–68. |
| [2] | E. Kosower, J. Cotter, Stable free radicals. Ⅱ. The reduction of 1-methyl-4-cyanopyridinium ion to methylviologen cation radical, J. Am. Chem. Soc. 86 (1964) 5524–5527. |
| [3] | R. Bozio, I. Zanon, A. Girlando, C. Pecile, Vibrational spectroscopy of molecular constituents of one-dimensional organic conductors. Tetrathiofulvalene (TTF), TTF+, and (TTF+)2 dimer, J. Chem. Phys. 71 (1979) 2282–2293. |
| [4] | (a) A.Y. Ziganshina, Y.H. Ko, W.S. Jeon, K. Kim, Stable π-dimer of a tetrathiafulvalene cation radical encapsulated in the cavity of cucurbit[8]uril, Chem. Commun. (2004) 806–807; (b) Y.M. Zhang, Y. Chen, R.J. Zhuang, Y. Liu, Construction and radical cation stabilization of a supramolecular dyad by tetrathiafulvalene-modified b-cyclodextrin and cucurbit[7]uril, Supramol. Chem. 23 (2011) 372–378. |
| [5] | C.A. Christensen, L.M. Goldenberg, M.R. Bryce, J. Becher, Synthesis and electrochemistry of a tetrathiafulvalene (TTF)21-glycol dendrimer: intradendrimer aggregation of TTF cation radicals, Chem. Commun. (1998) 509–510. |
| [6] | C. Wang, S.M. Dyar, D. Cao, et al., Tetrathiafulvalene hetero radical cation dimerization in a redox-active[2]catenane, J. Am. Chem. Soc. 134 (2012) 19136–19145. |
| [7] | H. Spanggaard, J. Prehn, M.B. Nielsen, et al., Multiple-bridged bis-tetrathiafulvalenes: new synthetic protocols and spectroelectrochemical investigations, J. Am. Chem. Soc. 122 (2000) 9486–9494. |
| [8] | M. Hasegawa, K. Daigoku, K. Hashimoto, H. Nishikawa, M. Iyoda, Face-to-face dimeric tetrathiafulvalenes and their cation radical and dication species as models of mixed valence and p-dimer states, Bull. Chem. Soc. Jpn. 85 (2012) 51–60. |
| [9] | C. Li, X. Zhao, X. Gao, Q. Wang, Z. Li, Foldamer-derived preorganized bi- and trizinc porphyrin tweezers for a pentafluorobenzene-bearing pyridine guest: the binding pattern study, Chin. J. Chem. 31 (2013) 582–588. |
| [10] | Z. Shi, Y. Song, F. Lu, et al., Evaluation on the stability of the intramolecular N– H…OMe hydrogen bonds of aromatic amide foldamers, Huaxue Xuebao 71 (2013) 51–61. |
| [11] | W.K. Wang, Y.Y. Chen, H. Wang, et al., Tetrathiafulvalene-based macrocycles formed by radical cation dimerization: the role of intramolecular hydrogen bonding and solvent, Chem. Asian J. 9 (2014) 1039–1044. |
| [12] | K.I. Nakamura, T. Hashimoto, T. Shirahata, et al., Synthesis and properties of new trimeric and tetrameric tetrathiafulvalenes with alternate links, Chem. Lett. 40 (2011) 883–885. |
| [13] | E. Gomar-Nadal, L. Mugica, J. Vidal-Gancedo, et al., Synthesis and doping of a multifunctional tetrathiafulvalene-substituted poly(isocyanide), Macromolecules 40 (2007) 7521–7531. |
| [14] | R. Kannappan, C. Bucher, E. Saint-Aman, et al., Viologen-based redox-switchable anion-binding receptors, New J. Chem. 34 (2010) 1373–1386. |
| [15] | A. Iordache, M. Oltean, A. Milet, et al., Redox control of rotary motions in ferrocene-based elemental ball bearings, J. Am. Chem. Soc. 134 (2012) 2653– 2671. |
| [16] | M.R. Geraskina, A.T. Buck, A.H. Winter, An organic spin crossover material in water from a covalently linked radical dyad, J. Org. Chem. 79 (2014) 7723–7727. |
| [17] | L.C. Cao, M. Mou, Y. Wang, Hyperbranched and viologen-functionalized polyglycerols: preparation, photo- and electrochromic performance, J. Mater. Chem. 19 (2009) 3412–3418. |
| [18] | K. Nchimi-Nono, P. Dalvand, K. Wadhwa, et al., Radical-cation dimerization overwhelms inclusion in[n]pseudorotaxanes, Chem. Eur. J. 20 (2014) 7334–7344. |
| [19] | L. Chen, H. Wang, D.W. Zhang, Y. Zhou, Z.T. Li, Quadruple switching on pleated foldamers of tetrathiafulvalene-bipyridinium-alternating dynamic covalent polymers, Angew. Chem. Int. Ed. (2015), http://dx.doi.org/10.1002/anie.201410757. |
| [20] | J.M. Spruell, Molecular recognition and switching via radical dimerization, Pure Appl. Chem. 82 (2010) 2281–2294. |
| [21] | S.V. Rosokha, J.K. Kochi, Molecular and electronic structures of the long-bonded π-dimers of tetrathiafulvalene cation-radical in intermolecular electron transfer and in (solid-state) conductivity, J. Am. Chem. Soc. 129 (2007) 828–838. |
| [22] | P.T. Chiang, N.C. Chen, C.C. Lai, S.H. Chiu, Direct observation of mixed-valence and radical cation dimer states of tetrathiafulvalene in solution at room temperature: association and dissociation of molecular clip dimers under oxidative control, Chem. Eur. J. 14 (2008) 6546–6552. |
| [23] | J. Tian, Y.D. Ding, T.Y. Zhou, et al., Self-assembly of three-dimensional supramolecular polymers through cooperative tetrathiafulvalene radical cation dimerization, Chem. Eur. J. 20 (2014) 575–584. |
| [24] | C. Zhou, J. Tian, J.L. Wang, et al., A three-dimensional water-soluble supramolecular organic framework stabilized by the dimerization of viologen radical cation, Polym. Chem. 5 (2014) 341–345. |
| [25] | L. Zhang, T.Y. Zhou, J. Tian, et al., Two-dimensional single-layer supramolecular organic framework that is driven by viologen radical cation dimerization and further promoted by cucurbit[8]uril, Polym. Chem. 5 (2014) 4715–4721. |
| [26] | Y.C. Zhang, Y. Zhou, Z.T. Li, D.W. Zhang, Stacking of hydrazone-bridged linear tetrathiafulvalene radical cations, Tetrahedron 71 (2015) 605–609. |
| [27] | Y. Wang, M. Frasconi, W.G. Liu, et al., Folding of oligoviologens induced by radical–radical interactions, J. Am. Chem. Soc. 137 (2015), http://dx.doi.org/10.1021/ja5111305. |
| [28] | L. Chen, S.C. Zhang, H. Wang, et al., Three-dimensional supramolecular polymers driven by rigid tetrahedral building blocks through tetrathiafulvalene radical cation dimerization, Tetrahedron 70 (2014) 4778–4783. |
| [29] | T.Q. Wan, Z.T. Li, From supramolecular polymers to supramolecular organic frameworks: engineering the periodicity of solution-phase self-assembled architectures, Imaging Sci. Photochem. 33 (2015) 3–14. |
| [30] | (a) M. Sun, H. Zhang, X. Hu, B. Liu, Y. Liu, Hyperbranched supramolecular polymer of tris(permethyl-β-cyclodextrin)s with porphyrins: characterization and magnetic resonance imaging, Chin. J. Chem. 32 (2014) 771–776; (b) L.H. Wang, Z.J. Zhang, H.Y. Zhang, H.L. Wu, Y. Liu, A twin-axial[5]pseudorotaxane based on cucurbit[8]uril and a-cyclodextrin, Chin. Chem. Lett. 24 (2013) 949–952. |
| [31] | F. Sakai, Z.W. Ji, J.H. Liu, G.S. Chen, M. Jiang, A novel supramolecular graft copolymer via cucurbit[8]uril-based complexation and its self-assembly, Chin. Chem. Lett. 24 (2013) 568–572. |
| [32] | Z.G. Tao, T.G. Zhan, T.Y. Zhou, X. Zhao, Z.T. Li, Synthesis, properties, and selfassembly of 2, 3-bis(n-octyl)hexaazatriphenylene, Chin. Chem. Lett. 24 (2013) 453–456. |