




b College of Chemical and Pharmaceutical Engineering, Hebei University of Science and Technology, Shijiazhuang 050018, China
β-lactamase production represents the most common form of antibiotic resistance [1, 2]. Four β-lactamase classes,A-D,have been identified. Classes A,C,and D are serine enzymes,whereas class B is zinc metalloenzyme [3]. Both classes A and C are clinically relevant [4]. Co-administration of antibiotics and a β-lactamase inhibitor has shown to be an effective strategy in overcoming antibiotic resistance [5]. Commercially available inhibitors,such as clavulanic acid,tazobactam,and sulbactam,inactivate most class A β-lactamases,but are ineffective against class C β-lactamases [6]. Recently,the incidence of infections by class C producing organisms has increased [7].
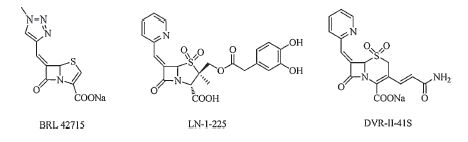
Introduction of an electron-withdrawing group,such as a double bond,into the β-lactam ring of a β-lactam antibiotic might increase its reactivity. It is hypothesized that the enhanced reactivity can extend the inhibitory activity to both class A and class C enzymes. This effect has been observed in several compounds,including BRL42715 [8],LN-I-255 [9],and DVR-Ⅱ- 41S [4] (Fig. 1). In 1980,Gordon et al. [10] reported that some 7-sulfenimine cephalosporins inhibit cephalosporinase. Furthermore,Changov [11] reported that penam sulfoxides showed the ability to inhibit β-lactamase. Based on these data, we synthesized a series of novel sulfenimine cephalosporin sulfoxide analogs and evaluated their ability to inhibit blactamase. The sulfenimine moiety,a strong electron-withdrawing group,can increase the reactivity of the β-lactam ring,and aid the inhibitors in reacting more readily with the active site of blactamases than β-lactam antibiotics. Furthermore,inhibitors maintaining a similar structure to β-lactam antibiotics can be easily recognized by β-lactamases. Thus they may have improved activity and selectivity.

|
Download:
|
| Fig. 1.Structures of β-lactamase inhibitors. | |
The synthesis of compounds (7a-v) is outlined in Scheme 1. To a suspension of 3-substituted cephalosporanic acid 3a-b (0.05 mol) and para-toluenesulfonic acid (9.5 g,0.05 mol) in water (25 mL) was added peroxyacetic acid (15%,27.9 g,0.055 mol) dropwise at 0℃ and the mixture was allowed to stir for 1 h. The reaction solution was adjusted to pH 2.5-3.0 by the addition of saturated sodium bicarbonate. The precipitates were collected by filtration and washed with water and acetone to obtain the key precursor compounds 4a-b (48.8%-64.5%).

|
Download:
|
| Scheme 1.Reagents and conditions: (a) Br2, DCM, -20℃, 30 min, (b) CH3CO3H, TsOH, H2O, 0℃, 1 h, (c) bis(trimethylsilyl)acetamide, DCM, reflux, 2 h, (d) corresponding sulfenyl bromide, DCM, 10℃, over 3 h, (e) H2O, r.t. | |
A mixture of substituted disulfide 1a-k (7.8 mmol) in dry methylene chloride (8 mL) was cooled to -20℃,and bromine (1.20 g,7.5 mmol) in methylene chloride (4 mL) was added dropwise over 10 min. The reaction mixture was stirred for 30 min at -20 ℃ to give the sulfenyl bromides 2a-k in methylene chloride.
A mixture of substituted cephalosporanic acid sulfoxide 4a-b (5 mmol),dry methylene chloride (15 mL),and bis(trimethylsilyl)[5TD$DIF]- acetamide (2.03 g,10 mmol) was heated to reflux for 2 h. The reaction mixture was[6TD$DIF] then cooled to 5℃,and propylene oxide (2.32 g,40 mmol) was added. A solution of sulfenyl bromides 2a-k was added dropwise while maintaining an inner temperature of 10℃. The reaction mixture was stirred for 3 h. The resulting mixture was filtered,and the filtrate was concentrated under reduced pressure. Saturated sodium bicarbonate (20 mL) was added to the residue and the solution was extracted with ethyl acetate (20 mL×2). The aqueous phase was acidified with 1 mol/L HCl to pH 2-3 and extracted with ethyl acetate (20 mL × 2). The combined organic extracts were washed with brine,dried over sodium sulfate,and concentrated under reduced pressure to afford crude products. The crude products were washed with methanol, and dried to give pure products 7a-v (50.2%-62.4%). Purity of all compounds was assessed by ultra performance liquid chromatography (UPLC) and determined to be >95% pure. The structures were confirmed by 1H nuclear magnetic resonance (NMR) and mass spectrometry (MS).
The inhibitory activity of the compounds was tested in vitro against the class A β-lactamase TEM-1,which is derived from Escherichia coli,and the class C enzyme cephalosporinase,which is derived from Bacillus. β-lactamase inhibitory activity was determined spectrophotometrically using nitrocefin as a substrate,as described by Bush et al. [12]. Nitrocefin was used at a final concentration of 180 mmol/L for TEM-1 and 200 mmol/L for cephalosporinase. The inhibitory activity was expressed as the half maximal inhibitory concentration (IC50). Compounds were assayed at 6 different concentrations (0.00032-1 mmol/L). In each experiment,tazobactam was used as a positive control. The most potent compounds were further evaluated in 1:2 or 1:1combinations with cefradine against a variety of clinically isolated β-lactamase-producing bacterial strains.
3. Results and discussion
As shown in Table 1,compounds 7b-d,7n,and 7s were more active than tazobactam against the class C enzyme,cephalosporinase. Interestingly,these compounds showed no or weak inhibitory activity against the class A enzyme TEM-1,suggesting that the presence of the sulfenimine moiety enhanced only class C enzyme inhibition. In most cases (7b-k,7m-p,7s,7u-v,especially 7d and 7n),the compounds bearing a bulky aromatic group at the sulfenimine moiety were more active than those bearing a methyl group. These results demonstrated that the pocket in the vicinity of C7 of β-lactamase is large. Furthermore the introduction of a halogen atom (F,Cl,or Br) at the benzene ring dramatically decreased inhibitory activity,suggesting that halogen groups may not be tolerated in this region.
|
|
Table 1 β-lactamase inhibitory activity against class A (TEM-1) and class C (cephalosporinase) enzymes. |

Table 2 shows the in vitro activity of 7c and 7n alone or in combination in 1:2 or 1:1 ratio with cefradine against clinically isolated β-lactamase-producing bacterial strains. The inhibitors alone had no antibiotic activity. However,combination therapy with cefradine synergistically inhibited (2-4 fold) several bacterial strains,particularly methicillin-sensitive Staphylococcus aureus (MSSA) and Klebsiella pneumoniae.
|
|
Table 2 In vitro synergy study of compounds 7c and 7n in combination with cefradine (CE) against clinically isolated β-lactamase-producing strainsa. |

{kind=link}
{kind=link}
In summary,the sulfenimine moiety could be used to enhance the inhibitory activity of cephalosporin sulfoxides against the class C enzyme cephalosporinase. The potency was significantly affected by the substituents on the sulfenimine moiety[1TD$DIF]. The study will provide valuable information for further research on the sulfenimine cephalosporin sulfoxide β-lactamase inhibitors.
Appendix A. Supplementary data
Supplementary data of representative compounds associated with this article can be found,in the online version,at http://dx.doi.org/10. 1016/j.cclet.2015.04.025.
| [1] | A. Copar, T. Prevec, B. Anžič, et al., Design, synthesis and bioactivity evaluation of tribactam β-lactamase inhibitors, Bioorg. Med. Chem. Lett. 12 (2002) 971-975. |
| [2] | X. Yang, Y.J. Zhou, P. He, et al., Activation free energy of Zn(Ⅱ), Co(Ⅱ) binding to metallo-β-lactamase ImiS, Chin. Chem. Lett. 25 (2014) 1323-1326. |
| [3] | S.M. Drawz, R.A. Bonomo, Three decades of β-lactamase inhibitors, Clin. Microbiol. Rev. 23 (2010) 160-201. |
| [4] | J.D. Buynak, L. Vogeti, V.R. Doppalapudi, G.M. Solomon, H.S. Chen, Cephalosporinderived inhibitors of β-lactamase. Part 4: The C3 substituent, Bioorg. Med. Chem. Lett. 12 (2002) 1663-1666. |
| [5] | T. Muratani, E. Yokota, T. Nakane, E. Inoue, S. Mitsuhashi, In-vitro evaluation of the four β-lactamase inhibitors: BRL42715, clavulanic acid, sulbactam, and tazobactam, J. Antimicrob. Chemother. 32 (1993) 421-429. |
| [6] | E.B. Grant, D. Guiadeen, E.Z. Baum, et al., The synthesis and SAR of rhodanines as novel class C β-lactamase inhibitors, Bioorg. Med. Chem. Lett. 10 (2000) 2179-2182. |
| [7] | J.D. Buynak, V.R. Doppalapudi, G. Adam, The synthesis and evaluation of 3-substituted-7-(alkylidene)cephalosporin sulfones as β-lactamase inhibitors, Bioorg. Med. Chem. Lett. 10 (2000) 853-857. |
| [8] | K. Coleman, D.R.J. Griffin, J.W. Page, P.A. Upshon, In vitro evaluation of BRL42715, a novel β-lactamase inhibitor, Antimicrob. Agents Chemother. 33 (1989) 1580-1587. |
| [9] | J.D. Buynak, A.S. Rao, V.R. Doppalapudi, et al., The synthesis and evaluation of 6-alkylidene-2' β-substituted penam sulfones as β-lactamase inhibitors, Bioorg. Med. Chem. Lett. 9 (1999) 1997-2002. |
| [10] | E.M. Gordon, H.W. Chang, C.M. Cimarusti, B. Toeplitz, J.Z. Gougoutas, Sulfenyl transfer rearrangement of sulfenimines (thiooximes). A novel synthesis of 7α-methoxycephalosporins and 6a-methoxypenicillins, J. Am. Chem. Soc. 102 (1980) 1690-1702. |
| [11] | L.S. Changov, E.D. Naydenova, G.I. Ivanova, et al., Synthesis and β-lactamase inhibitory activity of new 6β-cysteinesulfonamidopenicillanic acids, Bioorg. Med. Chem. 7 (1999) 2899-2904. |
| [12] | Y.J. Yang, R.T. Testa, N. Bhachech, et al., Biochemical characterization of novel tetrahydrofuranyl 1β-methylcarba-penems: stability to hydrolysis by renal dehydropeptidases and bacterial β-lactamases, binding to penicillin binding proteins, and permeability properties, Antimicrob. Agents Chemother. 43 (1999) 2904-2909. |