




Bedaquiline (Sirturo),which inhibits the proton pump for M. tuberculosis's ATP synthase,was developed by Johnson & Johnson and represented the first drug with a novel structure and mechanism for pulmonary MDR-TB in over 40 years [1, 2, 3]. To date, several synthetic methods for Bedaquiline have been reported. The original process for industrial preparation was disclosed by Koen Andries’s team at Johnson & Johnson [4, 5] (Scheme 1),in which a mixture of diastereoisomers were prepared in five steps and the ratio of the diastereoisomers (RS,SR)/(RR,SS) was 40/60. Subsequently Bedaquiline was isolated by a chiral resolution process in an overall yield of 6%. Later,Saga et al. [6] disclosed the first asymmetric synthesis of Bedaquiline in 12 steps in an overall yield of 5% using two key catalytic transformations: A catalytic enantioselective proton migration reaction using a bimetallic Ycomplex ligand and a CuF-catalyzed diastereoselective allylation reaction. The high cost of the catalyst and non-scalable reaction conditions of this route preclude its use for large-scale production. Chandrasekhar et al. [7] reported a synthesis of (2S)-stereoisomer and (2R)-stereoisomer from 6-bromo-2-chloroquinoline-3-carbaldehyde in an overall yield of 12% in 14 steps including a Sharpless asymmetric epoxidation,a regioselective epoxide opening and a modified allylzinc bromide addition. However,the above two asymmetric syntheses are limited to laboratory production due to the complicated operations,high cost of material,waste disposal and harsh reaction conditions. In contrast,the original patent route is more cost effective and convenient that only requires cheap, readily available reagents and an efficient chiral resolution process. Therefore,this first route is still being utilized for the large scale industrial production of Bedaquiline.
However,in this route,a large percent of other three nonpharmaceutically acceptable isomers,(1R,2R)-7,(1S,2S)-7,(1S, 2R)-7,will be inevitably produced together with Bedaquiline. Direct dispose of these inactive isomers would cause significant waste and increase the unnecessary cost of production. Here,we present a highly efficient way to recycle these inactive isomers 7 into two important intermediates 3 and 4 for the synthesis of Bedaquiline under basic conditions.
2. ExperimentalAll reactions were performed under a nitrogen atmosphere using anhydrous techniques unless otherwise noted. 1H NMR (300 MHz) on a Varian Mercury 300 spectrometer was recorded in DMSO-d6 or CDCl3. Chemical shifts are reported in δ relative to the internal standard tetramethylsilane (TMS). All the reactions were monitored by thin-layer chromatography (TLC) analysis on precoated silica gel G plates at 254 nm under UV lamp and HPLC.
2.1. General procedure for base-catalyzed Csp3-Csp3 bond cleavage of inactive stereoisomes 7NaOH powder (210 mg,5.4 mmol) was added to a stirred solution of inactive stereoisomes 7 (500 mg,0.9 mmol) in anhydrous THF (20 mL),then the reaction mixture was stirred at room temperature for 90 min. The progress of the reaction was monitored by TLC and HPLC analyses. After the completion of the reaction,the mixture was filtered and the filtrate was treated with 0.5 mol/L HCl and diluted with ethyl acetate. The organic layer was separated and removed by vacuum and the resulting residue was recrystallized from CH3OH to give 3 as a white solid. The water layer was treated with saturated Na2CO3 (aq.) and extracted with ethyl acetate. The organic layer was separated and dried over anhydrous MgSO4,after which the drying agent was filtered off and the volatiles were removed under reduced pressure to give 4 as a colorless oil. 3: Mp: 82-83 ℃. 1H NMR (300 MHz,DMSO-d6): δ 7.74 (d,1H, J = 2.0 Hz),7.69 (d,1H,J = 8.9 Hz),7.61 (dd,1H,J = 8.9,2.0 Hz),7.48 (s,1H),7.30-7.33 (m,2H),7.26-7.20 (m,3H),4.08 (s,3H),4.02 (s, 2H). 4: 1H NMR (300 MHz,CDCl3): δ 8.57 (d,1H,J = 8.4 Hz),7.98 (d, 1H,J = 8.4 Hz),7.87 (d,2H,J = 7.3 Hz),7.65-7.40 (m,3H),3.24 (t, 2H,J = 7.3 Hz),2.81 (t,2H,J = 7.3 Hz),2.29 (s,6H).
3. Results and discussionBedaquiline and its other three stereoisomers were prepared using the patent route as shown in Scheme 1. A mixture of 5 containing four isomers was obtained in four steps. Further chiral separation was performed by spontaneous crystallization to give diastereoisomer A and diastereoisomer B. The desired (1R,2S) enantiomer (i.e. Bedaquiline) was isolated using (R)-(-)-BNP ACID as a resolving agent from the diastereoisomer mixture A.

|
Download:
|
| Scheme 1.The industrial synthesis procedure of Bedaquiline | |
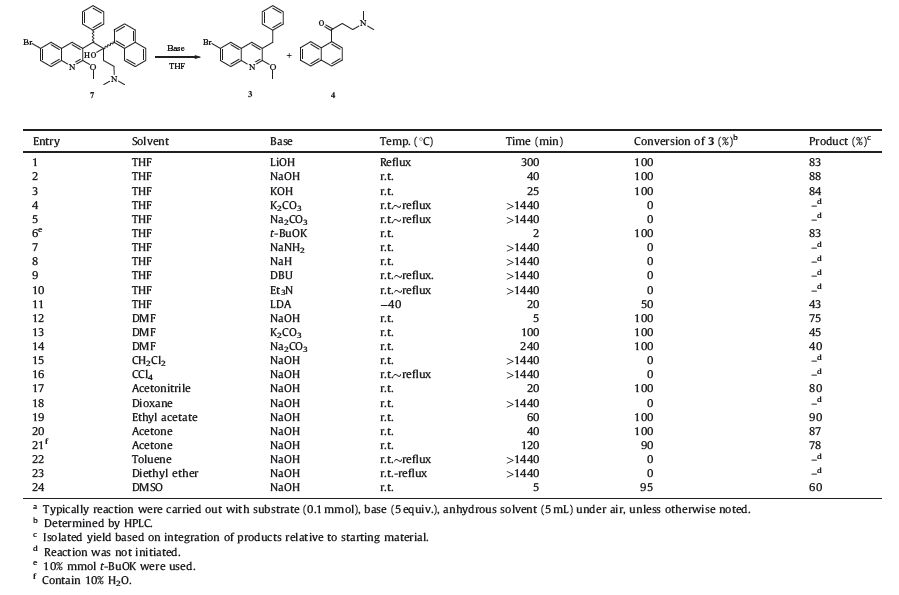
When inactive stereoisomes 7 was treated with sodium hydroxide in THF at room temperatures,the C-C bond between the two benzylic carbon atoms was cleaved,resulting in the formation of 3 and the corresponding ketone 4. This finding implies that the recovery of inactive stereoisomes can be achieved using a simple method. This reaction appeared to be a retro-aldol reaction, likely driven by a combination of the highly strained steric environment in the crowded carbinol 7,the acidity of 3,and the special proximity of the dimethyl amino group to the hydroxyl group in 7. So we speculated that the progress of the decomposition reaction may depend on the solvent and the base employed. Three factors,namely solvent,base,and temperature were selected to evaluate the decomposition step and the results are summarized in Table 1.
|
|
Table 1 Impact of reaction parameters on the direct decomposition of 7.a |
{kind=link}
After an initial screening of diffident conditions,we found that the base and solvent are both crucial for this decomposition reaction. Relative mild bases such as NaOH,KOH,LiOH could lead to the cleavage of 7 in THF in comparably high yields (Table 1, entries 1-3). However,weaker bases,such as K2CO3 and Na2CO3 (Table 1,entries 4 and 5),stronger bases,such as NaH and NaNH2 (Table 1,entries 7 and 8),as well as amine bases (Table 1,entries 9 and 10),were all completely ineffective when other conditions were the same as those shown in entry 2. In contrast,the strong base LDA at low temperature was found to be less effective with a large amount of byproducts produced and only a low yield of desired products was achieved (Table 1,entry 11). Notably,using catalytic amount of t-BuOK was proved to be more active than other bases,furnishing products in 83% yield (Table 1,entry 6). Replacing THF with other solvents such as ethyl acetate,acetone, acetonitrile in the presence of NaOH,can also result in satisfactory yields (>80%) (Table 1,entries 17,19 and 20). However,reactions in dichloromethane,carbon tetrachloride,dioxane,toluene and diethyl ether were not initiated completely (Table 1,entries 15,16, 18,22 and 23) and shows a much slower rate in acetone (containing 10% H2O) compared to anhydrous acetone (Table 1, entry 21). As shown in entries 12-14,weaker bases K2CO3 and Na2CO3 could not lead to the decomposition of substrate in THF, but this could be achieved in DMF. However,the reaction mixture was quite complicated. These results revealed that DMF could accelerate the reaction but produced more by-products. Similar results were also founded in DMSO (Table 1,entry 24). When considering the applicability in the scale-up and costs,catalytic amount of t-BuOK may be the best choice.
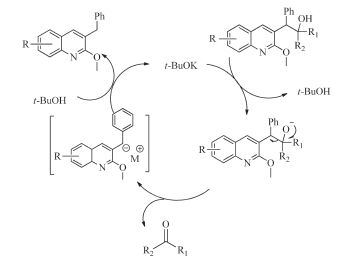
A tentative mechanism for the carbon-carbon bond cleavage is proposed in Scheme 2. Firstly,bases may remove the proton of hydroxyl and then the C-C bond between two benzylic carbon atoms was cleaved to form the corresponding ketone 4 and benzyl carbanion. Subsequently,carbanion will be protonated to give the other products 3.

|
Download:
|
| Scheme 2.Proposed mechanism for the base-catalyzed cleavage of 7. | |
{kind=link}
In summary,we have developed a simple,inexpensive,efficient method to recycle the inactive stereoisomers of Bedaquiline. The process described here can be utilized for the large scale preparation of Bedaquiline at low production costs. To date,the carbon-carbon bond cleavage of Bedaquiline and its stereoisomers catalyzed by bases has never been reported. Notably,unlike the retro-aldol reaction,the carbon-carbon bond cleavage between two benzylic carbons has only been sporadically studied [8]. After exhaustive evaluation of different reaction parameters,we have discovered the most proper bases for promoting the decomposition of Bedaquiline and its stereoisomers. Our study will provide new insights for further industrial preparation of drugs that have similar structures.
AcknowledgmentThis research was financially supported by the National Science and Technology Major Project of China (No. 521042).
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.04.013.
| [1] | E. Cox, K. Laessig, FDA approval of bedaquiline-the benefit-risk balance for drugresistant tuberculosis, N. Engl. J. Med. 371 (2014) 689-691. |
| [2] | R.V. Patel, S.D. Riyaz, S.W. Park, Bedaquiline: a new hope to treat multi-drug resistant tuberculosis, Curr. Top Med. Chem. 14 (2014) 1866-1874. |
| [3] | C.U. Köser, B. Javid, K. Liddell, et al., Drug-resistance mechanisms and tuberculosis drugs, Lancet 385 (2015) 305-307. |
| [4] | (a) G. Jerome Emile Georges, V. Gestel, J.F. Elisabetha, et al., Quinoline derivatives and their use as mycobacterial inhibitors, PCT Int. Appl. (2004), WO2004011436A1; (b) P. Frank Ralf, H. Stefan, B. Thomas, Process for preparing (aS, bR)-6-bromo-a-[2-(dimethylamino)ethyl]-2-methoxy-a-1-naphthaleny-b-phenyl-3-quiolineethanol, PCT Int. Appl. (2006), WO2006125769. |
| [5] | N. Lounis, J. Guillemont, N. Veziris, et al., R207910 (TMC207): a new antibiotic for the treatment of tuberculosis, Méd. Mal. Infect. 40 (2010) 383-390. |
| [6] | Y. Saga, R. Motoki, S. Makino, et al., Catalytic asymmetric synthesis of R207910, J. Am. Chem. Soc. 132 (2010) 7905-7907. |
| [7] | S. Chandrasekhar, G.S. Kiran Babu, D.K. Mohapatra, Practical syntheses of (2S)-R207910 and (2R)-R207910, Eur. J. Org. Chem. 11 (2011) 2057-2061. |
| [8] | P.J. Hamrick Jr., C.R. Hauser, The reversible addition of sodiodiphenylmethide to benzophenone in liquid ammonia, base-catalyzed cleavage of 1,1,2,2-tetraphenylethanol, J. Am. Chem. Soc. 81 (1959) 3144-3147. |