




b Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University, Shanghai 200062, China
Drug resistant Gram-negative infections have been a serious threat to public health,especially for hospitalized patients. Unfortunately,while resistance to current therapies is spreading rapidly,new antibacterial agents to treat these infections are few in number [1]. Thus,there is a great need to develop novel antibacterial agents with new mechanisms of action.
The outer membrane of the Gram-negative bacteria is a very efficient permeability barrier,obstructing the development of new antibiotics severely. Lipid A is a critical component of lipopolysaccharide (LPS),which mainly consists of the outer leaflet of the outer membrane [2]. The UDP-3-O-(R-3-hydroxyacyl)-N-acetylglucosamine deacetylase (LpxC) catalyzes the second-step of biosynthesis of lipid A in Gram-negative bacteria and is a validated antibiotic target [3, 4]. Thus,inhibition of LpxC can obstruct the biosynthesis of lipid A,and sequentially kill most Gram-negative bacteria [2].
The X-ray crystal structure of LpxC from Aquifex aeolicus (aaLpxC) reveals a catalytic zinc ion and a hydrophobic tunnel accommodating a myristate fatty acid side chain [5, 6]. Thus,most of the LpxC inhibitors contain a zinc-binding group (e.g. hydroxamic acid) and a hydrophobic tail which mimics the myristate fatty acid chain. These lipophilic moieties make important van der Waals interactions with the enzyme in the hydrophobic tunnel, which is critical for the inhibition of LpxC [1].
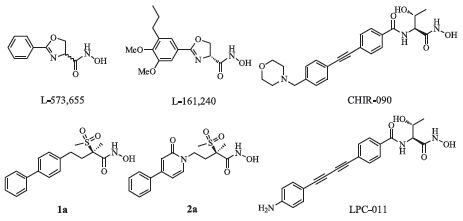
Since the discovery of L-573,655 and L-161,240 (Fig. 1) in 1996 [4],LpxC inhibitors have been developed for two decades. The inhibitors in the early days mainly exhibited strong antibacterial activities against Escherichia coli (E. coli),but rarely inhibited the growth of Pseudomonas aeruginosa (P. aeruginosa) [4, 7, 8, 9, 10, 11]. In 2004,Anderson et al. disclosed a new series of LpxC inhibitors, exemplified by CHIR-090 (Fig. 1),which displayed high affinity to both E. coli and P. aeruginosa LpxC and remarkable antibacterial activity against a wide range of Gram-negative pathogens [12]. On the basis of CHIR-090,Lee et al. [13] synthesized a series of compounds with linear diphenylacetylene scaffold which exhibited better antibacterial activities. In order to improve the watersolubility of these compounds,NH2 substituent was introduced in their distal phenyl ring,exemplified by LPC-011 (Fig. 1) [14]. LPC- 011 exhibited better water-solubility and antibacterial activity, while the metabolic data was not optimistic (the data will be mentioned later). In 2012,Pfizer reported a series of novel LpxC inhibitors with methylsulfone,exemplified by 1a (Fig. 1). 1a exhibited excellent antibacterial activity against P. aeruginosa but suffered from the problems of low water-solubility and high protein binding [1]. Then the central phenyl ring of 1a was replaced with pyridine (e.g. 2a in Fig. 1),and this series of compounds exhibited excellent Gram-negative antibacterial activity,improved solubility and increased free fraction. However,the half-life of these compounds in rats was shortened [15].

|
Download:
|
| Fig. 1.The structures of classic LpxC inhibitors. | |
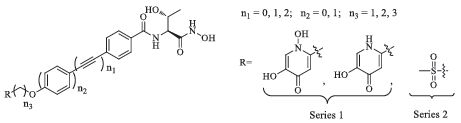
As discussed above,the balance of antibacterial activities and metabolic properties is a major challenge to the development of LpxC inhibitors. Thus,we designed and synthesized two series of compounds with hydrophilic terminus mainly aimed to achieve novel LpxC inhibitors with improved activity,metabolic stability and water solubility. The terminus of series 1 are kojic acid derivatives,which can deliver the compound through the outer membrane of the Gram-negative bacteria as a siderophore by the Trojan Horse approach [16]. Meanwhile,Kojic acid derivatives with NH3 and NH2OH substitution are proved effective siderophores [17, 18]. The terminus of series 2 is methylsulfone,which is a steady group for metabolism. Hydrophobic tails with different length have also been synthesized to match the hydrophobic tunnel (Fig. 2).

|
Download:
|
| Fig. 2.Two series of novel LpxC inhibitors. | |
The synthetic route of intermediates 4,7 and 10 was outlined in Scheme 1. Compound 2 was obtained by the coupling reaction of compound 1 and trimethylsilyacetyene in THF. The hydrolyzation and deprotection of compound 2 under alkaline condition afforded the acid compound 3,and then treated with L-threonine methyl ester hydrochloride in the condition of condensation reaction to afford compound 4. Protection of Kojic acid with 4-methoxybenzylchloride gave compound 5 and then treated with ammonium hydroxide to give compound 6. The reaction of compound 6 with 4-methoxybenzylchloride afforded the aromatization product compound 7. Compound 8 was obtained from the Kojic acid and diphenyldiazomethane under alkaline condition,then treated with alkalized hydroxylamine hydrochloride to yield compound 9. The reaction of compound 9 with diphenyldiazomethane afforded compound 10.

|
Download:
|
| Scheme 1.Conditions and reagents: (a) Trimethylsilylacetylene,PdCl2(PPh3)2,CuI,Et3N,THF,r.t.,98%; (b) NaOH,MeOH,r.t.,80%; (c) L-Threonine methyl ester hydrochloride, EDCI,HOBt,DIEA,DMF,r.t.,70%; (d) Kojic acid,PMBCl,K2CO3,DMF,80 ℃,80%; (e) NH3·H2O,EtOH,70 ℃,70%; (f) PMBCl,K2CO3,DMF,80 ℃,56.2%; (g) Ph2CN2,EtOH,40 ℃, 77%; (h) NH2OH·HCl,CH3COONa·3H2O,EtOH/H2O,70 ℃,44%; (i) Ph2CHCl,K2CO3,NaI,DMSO,r.t.,98%. | |
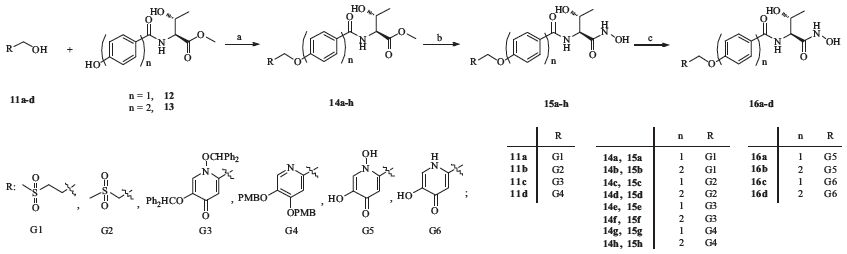
The synthetic route of compounds 15a-d and 16a-d was outlined in Scheme 2. Compounds 14a-h were obtained by the Mitsunobu reaction of RCH2OH and compounds 12 and 13,then treated with NH2OH (50% in water) and yielded compounds 15a-h. Deprotection of compounds 15e-h in trifluoroacetic acid yielded compounds 16a-d.

|
Download:
|
| Scheme 2.Conditions and reagents: (a) DEAD,PPh3,THF,r.t.,40%-60%; (b) NH2OH·H2O,DCM/MeOH,r.t.,40%-80%; (c) TFA,DCM,r.t.,70%-90%. | |
The synthetic route of compounds 22a-c and 23a-c was outlined in Scheme 3. Compounds 18a-d were obtained by the Mitsunobu reaction of RCH2OH and 4-iodoaniline. Compounds 19a and b were obtained by the coupling reaction of compounds 18a and d with trimethylsilyacetyene in THF,respectively. Then 19a and b were treated with tetrabutylammonium fluoride to yield compounds 20a and b. Compounds 21a-f were obtained by the coupling reaction of compound 4 with compounds 18a-d and 20a and b,then treated with NH2OH (50% in water) to yield compounds 22a-f. Deprotection of compounds 22d-f in trifluoroacetic acid yielded compounds 23a-c. Data of selected compounds can be found in supporting information.

|
Download:
|
| Scheme 3.Conditions and reagents: (a) DEAD,PPh3,THF,r.t.,40%-60%; (b) 4,PdCl2(PPh3)2,CuI,Et3N,THF,r.t.,80%-90%; (c) Trimethylsilylacetylene,PdCl2(PPh3)2,CuI,Et3N, THF,r.t.,80%-90%; (d) TBAF,THF,r.t.,40%-90%; (e) 4,Cu(Ac)2,Pyridine/MeOH,r.t.,20%-30%; (f) NH2OH·H2O,DCM/MeOH,r.t.,40%-80%; (g) TFA,DCM,r.t.,70%-90%. | |
Minimum inhibitory concentration (MIC) of each compound was tested by the modified National Committee for Clinical Laboratory Standards,which is adapted to 96-well plates and LB media in the presence of 5% DMSO [19]. Standards,which is adapted to tested bacterial strains were classic Gram-negative pathogens,including P. aeruginosa (PAO1,PA14) and E. coli (AB1157,DH5α). The MIC values of all tested compounds were listed in Table 1. Unfortunately,most compounds of series 1 lost their antibacterial activities. Only the compounds 23a and 23c with diphenylacetylene scaffold exhibited weak antibacterial activities against E. coli and P. aeruginosa. In series 2,compounds with biphenyl scaffold (15b) and diphenylacetylene scaffold (22b and 22c) showed remarkable antibacterial activities against both E. coli and P. aeruginosa. Especially,compounds 22b and 22c exhibited comparable antibacterial activities to CHIR-090. In both series,the length of the hydrophobic tails also played an important role: too long (diacetylene scaffold) or too short (phenyl scaffold) would lead to a loss of antibacterial activity.
|
|
Table 1 In vitro antibacterial activities of synthetic compounds. |
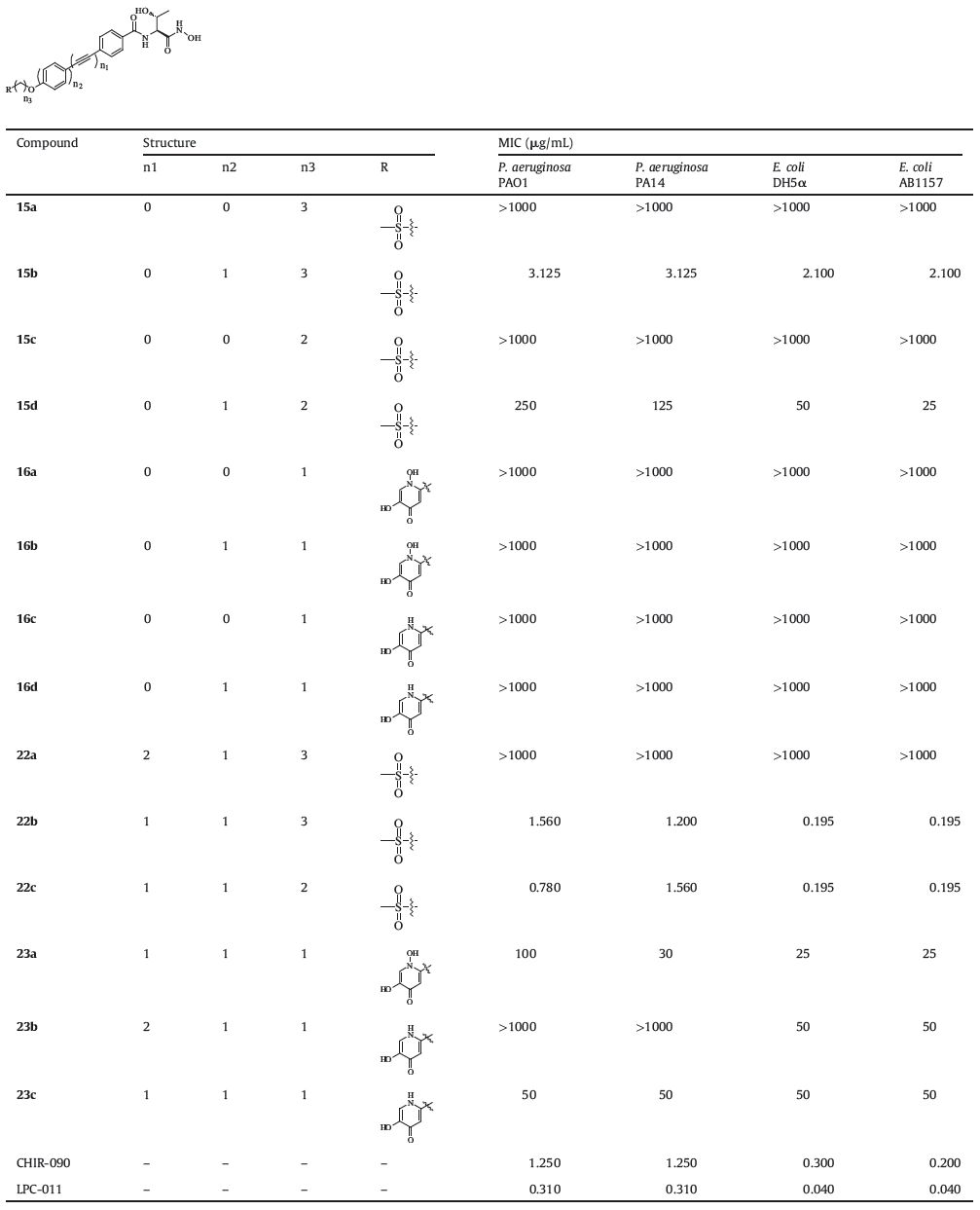
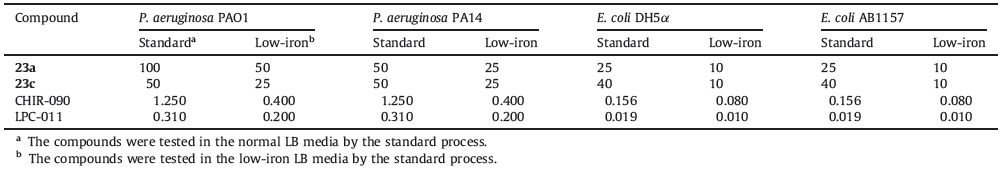
Moreover,we compared the antibacterial activities of compounds 23a and 23c,CHIR-090 and LPC-011 both under standard condition and low-iron condition. As shown in Table 2,all of the tested compounds exhibited better antibacterial activities under low-iron condition. Unexpectedly,similar improvements were observed in the antibacterial activities of tested compounds. It is speculated that the hydroxamic acid group can serve as a potent siderophore to help the inhibitors entering the outer membrane of the bacteria and the extra siderophore (kojic acid derivative) does not influence this process obviously. The detailed mechanism is not clear and needs further biological investigations.
|
|
Table 2 In vitro antibacterial activities of CHIR-090,LPC-011,23a and c under standard condition and low-iron condition. |
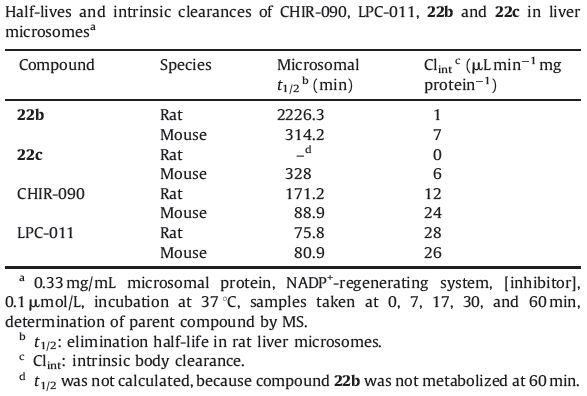
In addition,we evaluated the metabolic stability of compounds 22b and 22c,CHIR-090 and LPC-011 in liver microsomes (rat and mouse). The detail of the testing method can be found in supporting information. As shown in Table 3,compounds 22b and 22c exhibited better metabolic stability than CHIR-090 and LPC-011,which indicated that the terminal methylsulfone may be a preferred structure in the design of LpxC inhibitors.
|
|
Table 3 Half-lives and intrinsic clearances of CHIR-090,LPC-011,22b and 22c in liver microsomesa |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In conclusion,two series of novel LpxC inhibitors containing the terminal kojic acid derivatives and methylsulfone were synthesized and evaluated. The MIC values indicated that the length of the hydrophobic tail played an important role and the compounds with diphenylacetylene scaffold exhibited best antibacterial activities. In series 1,most of the compounds lost their antibacterial activities and the introduction of kojic acid derivatives did not provide extra improvement of antibacterial activities obviously compared to CHIR-090 and LPC-011 under low-iron condition. In series 2,some compounds,especially 22b and 22c,exhibited comparable antibacterial activities to CHIR-090 against E. coli and P. aeruginosa and better metabolic stability than CHIR-090 and LPC-011 in liver microsomes (rat and mouse). These results manifested the terminal methylsulfone may be a preferred structure in the design of LpxC inhibitors and worthy of further investigations.
AcknowledgmentsWe are grateful to National Science and Technology Major Project for the support of this research. The project described was supported by Key New Drug Creation and Manufacturing Program, China (No. 2014ZX09507009-016).
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.03.029.
| [1] | M.F. Brown, U. Reilly, J.A. Abramite, et al., Potent inhibitors of LpxC for the treatment of Gram-negative infections, J. Med. Chem. 55 (2012) 914-923. |
| [2] | T.J.O. Wyckoff, C.R.H. Raetz, J.E. Jackman, Antibacterial and anti-inflammatory agents that target endotoxin, Trends Microbiol. 6 (1998) 154-159. |
| [3] | A.W. Barb, A.L. McClerren, K. Snehelatha, et al., Inhibition of lipid A biosynthesis as the primary mechanism of CHIR-090 antibiotic activity in Escherichia coli, Biochemistry 46 (2007) 3793-3802. |
| [4] | H.R. Onishi, B.A. Pelak, L.S. Gerckens, et al., Antibacterial agents that inhibit lipid A biosynthesis, Science 274 (1996) 980-982. |
| [5] | D.A. Whittington, K.M. Rusche, H. Shin, C.A. Fierke, D.W. Christianson, Crystal structure of LpxC, a zinc-dependent deacetylase essential for endotoxin biosynthesis, Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 8146-8150. |
| [6] | B.E. Coggins, X.C. Li, A.L. McClerren, et al., Structure of the LpxC deacetylase with a bound substrate-analog inhibitor, Nat. Struct. Mol. Biol. 10 (2003) 645-651. |
| [7] | M.H. Chen, M.G. Steiner, S.E. de Laszlo, et al., Carbohydroxamido-oxazolidines: antibacterial agents that target lipid A biosynthesis, Bioorg. Med. Chem. Lett. 9 (1999) 313-318. |
| [8] | J.E. Jackman, C.A. Fierke, L.N. Tumey, et al., Antibacterial agents that target lipid A biosynthesis in gram-negative bacteria, J. Biol. Chem. 275 (2000) 11002-11009. |
| [9] | M.C. Pirrung, L.N. Tumey, C.R.H. Raetz, et al., Inhibition of the antibacterial target UDP-(3-O-acyl)-N-acetylglucosamine deacetylase (LpxC): isoxazoline zinc amidase inhibitors bearing diverse metal binding groups, J. Med. Chem. 45 (2002) 4359-4370. |
| [10] | P. Calí, L. Nærum, S. Mukhija, A. Hjelmencrantz, Isoxazole-3-hydroxamic acid derivatives as peptide deformylase inhibitors and potential antibacterial agents, Bioorg. Med. Chem. Lett. 14 (2004) 5997-6000. |
| [11] | J.M. Clements, F. Coignard, I. Johnson, et al., Antibacterial activities and characterization of novel inhibitors of LpxC, Antimicrob. Agents Chemother. 46 (2002) 1793-1799. |
| [12] | J. Zhang, L. Zhang, X. Li, W. Xu, UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC) inhibitors: a new class of antibacterial agents, Curr. Med. Chem. 19 (2012) 2038-2050. |
| [13] | C.J. Lee, X.F. Liang, X. Chen, et al., Species-specific and inhibitor-dependent conformations of LpxC: implications for antibiotic design, Chem. Biol. 18 (2011) 38-47. |
| [14] | X.F. Liang, C.J. Lee, X. Chen, et al., Syntheses, structures and antibiotic activities of LpxC inhibitors based on the diacetylene scaffold, Bioorg. Med. Chem. 19 (2011) 852-860. |
| [15] | J.I. Montgomery, M.F. Brown, U. Reilly, et al., Pyridone methylsulfone hydroxamate LpxC inhibitors for the treatment of serious gram-negative infections, J. Med. Chem. 55 (2012) 1662-1670. |
| [16] | U. Möllmann, L. Heinisch, A. Bauernfeind, T. Kö hler, D. Ankel-Fuchs, Siderophores as drug delivery agents: application of the "Trojan Horse" strategy, Biometals 22 (2009) 615-624. |
| [17] | M.G.P. Page, C. Dantier, E. Desarbre, In vitro properties of BAL30072, a novel siderophore sulfactamwith activity against multiresistant Gram-negative bacilli, Antimicrob. Agents Chemother. 54 (2010) 2291-2302. |
| [18] | M.E. Flanagan, S.J. Brickner, M. Lall, et al., Preparation, gram-negative antibacterial activity, and hydrolytic stability of novel siderophore-conjugated monocarbam diols, ACS Med. Chem. Lett. 2 (2011) 385-390. |
| [19] | X.F. Liang, C.J. Lee, J.S. Zhao, E.J. Toone, P. Zhou, Synthesis, structure, and antibiotic activity of aryl-substituted LpxC inhibitors, J. Med. Chem. 56 (2013) 6954-6966. |