




The α-amination of carbonyl compounds is one of the most important transformations in organic synthesis and numbers of nitrogen containing derivatives were successfully synthesized [1, 2, 3]. In particular,the enantioselective α-amination reaction,an important transformation that offers optically active α-amino acid derivatives,which are fundamental constituents of numerous natural products and pharmaceuticals,has attracted considerable attention in recent years [4, 5, 6, 7]. Several asymmetric variants have been reported [8, 9, 10, 11]. For example,Jørgensen and coworkers reported the first bisoxazoline-copper (II) complex catalyzed asymmetric α-amination of 2-ketoesters with azodicarboxylates [12]. Later,Jørgensen’s group developed the L-proline promoted asymmetric α-amination of ketone with azodicarboxylates providing the corresponding α-hydrazino ketones in up to 94% ee [13]. Almost at the same time,List and coworkers reported the (S)- proline catalyzed the enantioselective α-amination of unbranched aldehydes with dialkyl azodicarboxylates to give the α-aminoaldehydes in 95% ee [14]. In 2010,Rawalet al. developed the chiral squaramide organocatlyst derived from cyclohexane-1,2-diamine, which shown excellent catalytic performance in asymmetric α-hydrazination of 1,3-dicarbonyl compounds with azodicarboxylates [15]. More recently,Wang reported the amine-thiourea organocatalyst catalyzed the asymmetric amination of cyclic β-keto esters reaction [16]. Despite the progress has been made in recent years,the enantioselctive α-amination reaction catalyzed by BINOL-squaramides has been rarely described. However,there is still room for improvement regarding this type of asymmetric reactions; the existing catalytic system is still limited.
As part of our research on asymmetric catalysis reaction [17, 18, 19, 20, 21],we became interested in applying the chiral BINOLsquaramide as organocatalyst in asymmetric α-amination reaction of 1,3-dicarbonyl with dialkyl azodicarboxylates. Given that the chiral squaramide derivatives can function as highly efficient multiple H-bond donor organocatalysts in variety of asymmetric transformations [22, 23, 24, 25, 26, 27],we began to examine other reactions to further assess the enantioselectivity of this kind of chiral squaramide organocatalyst.
Herein,we describe the BINOL-quinine-squaramide as a highly efficient catalyst for the enantioselective amination of 1,3- dicarbonyl compounds with azodicarboxylates,which afforded valuable chiral amino acids derivatives in high yields and excellent enantioselectivities (up to 98% ee).
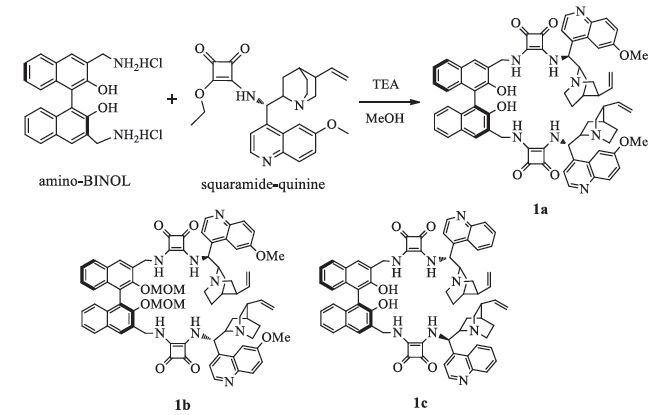
First,the chiral organocatalysts 1a (Scheme 1) were prepared in 75% yield through one step from easily available amino-BINOL and quinine according to previous method [17]. For comparison,the 1b and 1c were also synthesized through the same methods in 78% and 69% yields,respectively (Scheme 1).

|
Download:
|
| Scheme 1.Synthesis of chiral squaramides organocatalysts. | |
Analytical-grade solvents were purchased and used as received. NMR spectra were measured at 400 MHz for 1H NMR and 100 MHz for 13C NMR and calibrated from residual solvent signal. Analytical thin-layer chromatography (TLC) was performed on silica gel aluminum sheets with F-254 indicator. Visualization was accomplished by UV light. Mass spectra (MS) were measured on IonSpec 4.7 Tesla FTMS using DART Positive. Purification by chromatography was performed using 230-400 mesh SiO2 with compressed air as a source of positive pressure. The data of these known products 4a-d [28],4e-h [29],4m [30],4n [28],were consistent with those reported in the corresponding references.
2.1. Representative procedure for the synthesis of catalysts 1aA solution of amino-binaphthol (0.1 mmol) in MeOH (5 mL) with TEA (0.25 mmol) was stirred at room temperature for 30 min, then the quinine-squaramide (0.3 mmol) was added. The mixture was stirred for another 48 h. The precipitate was filtered and the ultimate product was recrystallized from MeOH to afford the product 1a in 75% yield as a yellow solid.
1a: Yellow solid,mp: 236 ℃ decomposed,[α]D25 -75 (c 0.54,DMSO),IR (KBr,cm-1): 3389,3234,2936,1797,1621,1584,1473,1434,1385,1357,1263,1225,1144,1099,1028,849,780. 1H NMR (400 MHz,DMSO-d6): δ 8.81 (s,2H),8.13 (s,2H),7.99 (d,2H, J= 9.2 Hz),7.83 (s,8H),7.59 (d,2H,J= 2.4 Hz),7.45 (d,2H, J= 7.6 Hz),7.22 (t,2H,J= 8.0 Hz),7.11 (t,2H,J= 8.0 Hz),6.78 (d,2H,J= 8.0 Hz),6.00 (s,4H),5.07-4.98 (m,8H),3.93 (s,6H),3.35 (s,6H),2.68 (s,4H),2.27 (s,2H),1.51 (m,8H),0.60 (s,2H). 13C NMR (100 MHz,DMSO-d6): δ182.74,182.20,167.93,167.05, 158.27,158.07,142.84,142.64,131.82,131.62,128.82,128.33, 127.87,126.3,124.54,123.22,122.40,119.56,114.77,101.93, 59.16,56.11,53.15,44.31,27.73,26.70. HRMS (ESI) calcd. for C70H66N8O8 [M+H]+ 1147.5082; found 1147.5076.
1b: Yellow solid,mp: 190-193 ℃,[α]D25- 36:9 (c 0.77,DMSO), IR (KBr,cm-1): 3241,3068,2936,1796,1670,1621,1587,1530, 1474,1347,1262,1149,1090,973,917,851,783. 1H NMR (400 MHz,CDCl3): δ8.99 (s,2H),8.76 (s,2H),7.91 (m,8H),7.71 (d,4H,J= 7.6 Hz),7.37 (m,4H),7.15 (m,4H),6.38 (s,2H),5.86 (s,2H), 5.00 (m,8H),4.04 (s,6H),4.01 (s,4H),3.64 (s,4H),3.35 (s,2H),3.03 (m,6H),2.86 (s,4H),2.31 (m,8H),1.51 (s,2H),0.73 (s,2H). 13C NMR (100 MHz,CDCl3): δ182.71,182.35,167.47,158.63,152.88, 148.10,144.62,143.67,140.80,133.64,131.51,130.61,129.69, 128.22,127.86,126.89,125.88,125.31,124.71,122.50,119.55, 115.23,101.20,99.25,77.22,60.18,56.40,56.39,56.40,46.32, 40.93,27.45,26.01. HRMS (ESI) calcd. for C74H74N8O10 [M+H]+ 1235.5606; found 1235.5601.
1c:Yellow solid,mp: 247 ℃ decomposed,[α]D25- 68:9 (c 0.60,DMSO),IR (KBr,cm-1): 3423,3062,2958,1798,1675,1601,1530,1459,1391,1359,1260,1241,1146,1105,1035,813,767,752. 1H NMR (400 MHz,DMSO-d6): δ10.14 (s,2H),9.02 (s,4H),8.45 (d,6H, J= 8.0 Hz),8.11 (d,2H,J= 8.0 Hz),7.82 (m,8H),7.21 (m,6H),6.31 (s,2H),6.01 (s,2H),5.15 (m,4H),4.89 (s,4H),4.59 (s,2H),3.92 (s, 2H),3.72 (s,2H),2.79 (s,2H),1.92 (m,8H),1.27 (s,2H),0.99 (s,2H), 0.86 (s,2H). 13C NMR (100 MHz,DMSO-d6): δ182.79,169.74,166.81,152.05,150.95,148.58,142.76,142.56,138.91,133.88, 130.41,128.95 127.98,126.38,126.17,124.40,123.35,119.87,117.03,114.39,59.65,53.60,51.84,40.84,36.70,24.28,24.01.HRMS (ESI) calcd. for C68H62N8O6 [M+H]+ 1087.4871; found 1087.4865.
2.2. General procedure for α-hydrazination reaction of 1,3-dicarbonyl compoundsIn a 10 mL round-bottomed flask,the 1,3-dicarbonyl compound 2 (0.24 mmol) was added to the agitated solution of catalyst 1a (0.01 mmol) in 1 mL THF at -78 ℃ and stirred for 15 min, compound 3 (0.20 mmol) in 1 mL THF was added. The reaction was monitored by TLC and condensed under reduced pressure and subjected to flash chromatography column to give the pure product 4.
4a: 1H NMR (400 MHz,CDCl3): δ6.81 (s,1H),4.17-4.10 (m,6H), 2.23-1.91 (m,6H),1.22-1.16 (m,9H). 13CNMR(100 MHz,CDCl3): d 155.92,155.56,63.05,62.35,29.63,24.95,18.58,14.35,14.24, 13.98. MS-ESI [M+H]+ m/z,331. Chiralpak AS-H column (250 mm × 4.6 mm),10% iPrOH/hexane; 0.9 mL/min,254 nm; Rtmajor = 16.750 min,Rtminor = 22.283 min,77% ee.
4b: 1H NMR (400 MHz,CDCl3): δ6.69 (s,1H),4.91 (t,2H,J= 5.2 Hz),4.23 (t,2H,J= 6.8 Hz),2.66-1.97 (m,6H),1.30-1.23 (m,15H). 13C NMR (100 MHz,CDCl3): δ206.68,204.78,167.97,155.78,155.04,71.08,69.90,62.15,53.10,36.23,33.30,32.10,30.79,21.72,18.57,13.90. Chiralpak AD-H column (250 mm × 4.6 mm),5% iPrOH/hexane; 1 mL/min,210 nm; Rtmajor = 11.572 min,Rtminor = 16.582 min,85% ee.
4c: 1H NMR (400 MHz,CDCl3): δ6.53 (s,1H),4.22 (m,2H),2.58- 1.95 (m,6H),1.45 (s,18H),1.29 (t,3H,J= 7.2 Hz). 13C NMR (100 MHz,CDCl3): δ155.22,154.26,82.47,81.22,77.51,77.19, 76.87,61.79,60.14,37.27,35.67,31.92,27.97,27.86,18.50,14.06. ESI [M]+ m/z,386. Chiralpak AD-H column (250 mm × 4.6 mm), 5% iPrOH/hexane; 1 mL/min,210 nm; Rtmajor = 8.293 min, Rtminor = 16.262 min,58% ee.
4d: 1HNMR(400 MHz,CDCl3): δ7.20 (m,10H),7.00 (s,1H),5.01 (s,4H),4.06 (s,2H),2.55-1.88 (m,6H),1.08 (t,3H). 13C NMR (100 MHz,CDCl3): δ156.03,135.44,135.23,128.60,128.54, 128.46,128.37,128.11,128.04,68.75,67.89,62.56,29.69,18.58, 13.82. ESI [M+H]+ m/z,455. Chiralpak AS-H column (250 mm × 4.6 mm),25% iPrOH/hexane; 0.7 mL/min,254 nm; Rtmajor = 18.355 min,Rtminor = 35.662 min,89% ee.
4e: 1H NMR (400 MHz,CDCl3): δ6.81 (s,1H),4.18 (m,4H),3.78 (s,3H),2.30 (m,6H),1.29-1.23 (m,6H). 13C NMR (100 MHz, CDCl3): δ156.13,155.54,63.11,62.34,53.13,36.62,31.72,18.58, 14.29,14.18. ESI [M+H]+ m/z,316. Chiralpak AD-H column (250 mm × 4.6 mm),5% iPrOH/hexane; 1 mL/min,210 nm; Rtmajor = 18.392 min,Rtminor = 25.507 min,90% ee.
4f: 1H NMR (400 MHz,CDCl3): δ6.67 (s,1H),5.06-4.78 (m,2H),3.75 (s,3H),2.30 (br,6H),1.25 (br,12H). 13C NMR (100 MHz,CDCl3): δ155.86,155.10,71.26,70.10,69.77,53.14,36.54,35.98, 32.03,21.79,18.61. ESI [M+H]+ m/z,345. Chiralpak AD-H column (250 mm × 4.6 mm),5% iPrOH/hexane; 1 mL/min,210 nm; Rtmajor = 4.977 min,Rtminor = 6.393 min,84% ee.
4g: 1H NMR (400 MHz,CDCl3): δ6.50 (s,1H),3.77 (s,3H),2.27 (m,6H),1.44 (m,18H). 13C NMR (100 MHz,CDCl3): δ167.73, 155.13,154.17,82.66,81.37,53.19,36.39,31.87,28.01,27.89, 27.81,18.34. ESI [M]+ m/z,372. Chiralpak AD-H column (250 mm × 4.6 mm),5% iPrOH/hexane; 1 mL/min,210 nm; Rtmajor = 14.578 min,Rtminor = 25.513 min,41% ee.
4h: 1H NMR (400 MHz,CDCl3): δ7.31-7.28 (m,10H),7.00 (s, 1H),5.12 (d,4H,J= 8.4 Hz),3.67 (s,3H),2.57-1.95 (m,6H). 13C NMR (100 MHz,CDCl3): δ156.04,135.36,135.20,128.60,128.55, 128.49,128.40,128.14,128.06,68.94,67.95,60.27,53.07,21.06, 18.83,14.14. ESI [M+H]+ m/z,441. Chiralpak AS-H column (250 mm × 4.6 mm),25% iPrOH/hexane; 0.7 mL/min,254 nm; Rtmajor = 22.845 min,Rtminor = 42.320 min,98% ee.
4i: [α]D25+ 5.2 (c 0.46,CHCl3),1H NMR (400 MHz,CDCl3): δ6.84 (s,1H),4.78 (s,2H),4.33-4.03 (m,4H),2.66 (s,2H),2.52 (s,1H), 2.39 (s,1H),2.23 (s,1H),2.00 (s,1H),1.79 (s,1H),1.26 (m,6H). 13C NMR (100 MHz,CDCl3): δ156.17,77.29,76.66,75.74,63.00,62.34, 53.40,29.72,18.35,14.36,14.24. ESI [M+H]+ m/z,340. Chiralpak AD-H column (250 mm × 4.6 mm),10% iPrOH/hexane; 1 mL/min, 210 nm; Rtmajor = 14.652 min,Rtminor = 17.498 min,94% ee.
4j: [α]D25+ 1.9 (c 0.37,CHCl3),1H NMR (400 MHz,CDCl3): δ6.70 (s,1H),5.01-4.87 (m,2H),4.75 (m,2H),2.65 (s,2H),2.51 (s,1H), 2.37 (s,1H),2.22 (s,1H),1.98 (s,1H),1.75 (s,1H),1.32-1.20 (m, 12H). 13C NMR (100 MHz,CDCl3): δ155.86,154.93,77.28,76.70, 75.74,71.49,70.22,53.53,21.84,18.66. ESI [M+H]+ m/z,369. Chiralpak AD-H column (250 mm × 4.6 mm),10% iPrOH/hexane; 1 mL/min,210 nm; Rtmajor = 9.488 min,Rtminor = 13.227 min, 87% ee.
4k: [α]D25- 3.4 (c 0.32,CHCl3),1H NMR (400 MHz,CDCl3): δ6.55 (s,1H),4.75 (s,2H),2.69 (s,2H),2.51 (s,1H),2.36 (s,1H),2.22 (s,1H), 2.00 (s,1H),1.85 (s,1H),1.45 (d,18H,J= 8.0 Hz). 13C NMR (100 MHz,CDCl3): δ155.19,154.23,82.99,81.59,77.27,76.83, 75.62,53.41,28.09,27.99,27.91,18.53. ESI [M]+ m/z,396. Chiralpak AD-H column (250 mm × 4.6 mm),10% iPrOH/hexane; 1 mL/min, 210 nm; Rtmajor = 10.050 min,Rtminor = 16.388 min,70% ee.
4l: [α]D25+ 6.4 (c 0.31,CHCl3),1H NMR (400 MHz,CDCl3): δ7.30 (s,10H),6.94 (s,1H),5.11 (d,4H,J= 8.4 Hz),4.67 (s,2H),2.70 (s,2H),2.45 (s,1H),2.35 (s,1H),2.21 (s,1H),1.99 (s,1H),1.64 (s,1H). 13C NMR (100 MHz,CDCl3): δ135.34,135.06,128.61, 128.57,128.48,128.42,128.06,77.30,76.63,75.83,68.85,67.97, 53.68,18.59. ESI [M+H]+ m/z,465. Chiralpak AS-H column (250 mm × 4.6 mm),30% iPrOH/hexane; 0.8 mL/min,254 nm; Rtmajor = 27.760 min,Rtminor = 49.998 min,92% ee.
4m: 1HNMR(400MHz,CDCl3): d6.76(s,1H),4.38-4.02 (m,4H), 2.58 (m,4H),2.28 (s,3H),2.04 (s,1H),1.88 (s,1H),1.27 (m,6H). 13C NMR (100MHz,CDCl3): δ156.29,155.89,82.36,63.44,62.42,60.31, 36.87,30.36,24.95,17.82,14.34,14.18. ESI [M+H]+ m/z,301.Chiralpak AD-H column (250mm× 4.6 mm),5% iPrOH/hexane; 1mL/min, 210 nm; Rtmajor = 22.222min,Rtminor = 27.197min,59% ee.
4n: 1H NMR (400 MHz,CDCl3): δ6.70 (s,1H),4.94 (m,2H),2.78 (s,1H),2.37 (s,2H),2.29 (s,3H),2.02 (s,2H),1.88 (s,1H),1.26 (s, 12H). 13C NMR (100 MHz,CDCl3): δ156.14,155.39,82.13,71.29,70.12,36.90,30.15,29.55,24.95,21.75,17.96,14.00. ESI [M+H]+ m/z,329. Chiralpak AD-H column (250 mm × 4.6 mm),5% iPrOH/ hexane; 1 mL/min,210 nm; Rtmajor = 8.927 min,Rtminor = 12.320 min,43% ee.
4o: [α]D25+ 15:9 (c 0.34,CHCl3),1H NMR (400 MHz,Acetone): δ7.31 (s,10H),7.05 (s,1H),5.12 (d,4H,J= 10 Hz),2.77 (s,1H),2.37 (s,2H),2.20 (s,3H),1.97 (s,1H),1.85 (s,2H). 13C NMR (100 MHz, CDCl3): δ171.48,156.95,135.03,128.62,128.58,128.53,128.17, 82.42,69.00,68.03,60.47,36.89,30.62,24.99,21.03,18.00,14.16. ESI [M+H]+ m/z,425. Chiralpak AS-H column (250 mm × 4.6 mm), 5% iPrOH/hexane; 1 mL/min,210 nm; Rtmajor = 29.808 min, Rtminor = 34.047 min,60% ee.
2.3. Catalyst recoveryAfter the reaction was finished,the mixture was centrifuged and the catalyst deposited at the bottom of the vial. The liquid layer was siphoned out and the residual solid was washed until no more compounds were detected by TLC. Then the vial with remaining catalyst was dried under vacuum.
3. Results and discussionWith the catalyst 1a-c in hand,their activity was tested in the addition of β-ketoester 2a with diethyl azodicarboxylate 3a. The results were summarized in Table 1. To our delight,the reaction was finished in 5 h at -60 ℃ in the presence of 5 mol% of catalyst 1a,and the desired product 4a was obtained in 95% yield and 77% ee (entry 1). Much lower enantioselectivity was observed by MOM-protected squaramide 1b (entry 2). For comparison,when the catalyst 1c was used in this reaction,4a was formed in 96% yield and 46% ee (entry 3). On the basis of observed reactivity of these catalysts,it clearly demonstrated that the amino and hydroxyl groups were both catalytic active sites in the catalytic cycle. The squaramide organocatalyst was assumed to create multiple hydrogen-bonding sites in which squaramide and cinchonine moiety interacted through hydrogen bonding with diketone simultaneously. Meanwhile,two OH groups of binaphthyl moiety provide hydrogen bonding to azodicarboxylate. The tertiary amine of cinchonine moiety plays the role of the base to deprotonate α-carbon of the carbonyl group,forming the enolate to attack the electrophile,which leads to the formation the products.
|
|
Table 1 Optimized condition for the reaction of 2a with 3a catalyzed by 1a-c |

The screening of solvents indicated that THF was the best solvent giving the product in 77% ee. Other solvents,such as toluene,DCM,CHCl3,DMF and methanol gave lower ee values (entries 4-8). When the catalyst loading was reduced from 5 mol% to 1 mol%,the enantioselectivity was slightly decreased. Reducing the reaction temperature to -78 ℃ led to improvement of enantioselectivity affording the desired product in 85% ee (entry 11).
Under the optimized reaction conditions,we next investigated the scope and limitation of the enantioselective α-amination of 1,3-dicarbonyl compounds with dialkyl azodicarboxylates,and the result were summarized in Table 2. As illustrated in Table 2,the Michael addition underwent cleanly to give the desired adducts 4 in full conversion and up to 98% ee in the presence of 5 mol% BINOL-quinine-squaramide 1a (entries 1-15). Azodicarboxylates 3 bearing more bulky groups reacted smoothly with 1,3-dicarbonyl compound 2,exclusively affording the corresponding products in excellent ee values (entries 4,8,12). In particular,the reaction of methyl 2-oxocyclopentanecarboxylate 2b with azodicarboxylate 3d afforded the corresponding adduct 4h with the highest enantioselectivity (98% ee) (entry 8). While,when 2-acetylcyclopentanone 2d was used in this amination reaction,the products 4m-o were formed in lower ee values (entries 13-15). The zaodicarboxylate bearing the most steric t-Bu group exhibited lower reactivity and enantioselectivity (entry 3),which may attribute to the steric effects.
|
|
Table 2 Asymmetric Michael addition of 1,3-dicarbonyl compounds 2 with 3 catalyzed by 1aa. |

{kind=link}
In conclusion,we have developed an organocatalytic enantioselectivitive α-hydrazination reaction of 1,3-dicarbonyl compound with dialkyl azodicarboxylates. The reaction proceeds well in the presence of C2-symmetric BINOL-squaramide providing the desired products in high yield and in up to 98% ee. These result significantly expanded the scope of the asymmetric catalysis of bifunctional squaramide. Future investigation of those bifunctional organocatalyst in other asymmetric reaction is ongoing in our laboratory and will be reported in due course.
AcknowledgmentWe are grateful to the Fundamental Research Funds for the Central Universities (No. 2042014kf0248) for support of this research.
| [1] | J.P. Genet, C. Greck, D. Lavergne, in: A. Ricci (Ed.), Modern Amination Methods, Wiley-VCH, Weinheim, Germany, 2000. |
| [2] | (a) G. Boche, in: Helmchen (Ed.), Stereoselective Synthesis, vol. 9, Thieme, Stuttgart, Germany, 1996, pp. 5133-5157; (b) T.C. Li, C.X. Chen, X.Q. Li, X.H. Gao, Selective amination of the nucohalic acid derivatives, Chin. Chem. Lett. 24 (2013) 202-204. |
| [3] | H. Gröger, Catalytic enantioselective strecker reaction and analogus synthesis, Chem. Rev. 103 (2003) 2795-2828. |
| [4] | A. Círdova, The direct catalytic asymmetric Mannich reaction, Acc. Chem. Res. 37 (2004) 102-112. |
| [5] | H.R. Kricheldorf, Polypeptides and 100 years of chemistry of a-amino acid N-carboxyanhydrides, Angew. Chem. Int. Ed. 45 (2006) 5752-5784. |
| [6] | P.M. Pihko, A. Pohjakallio, Enantioselective oganocatalytic amination: a-aminations of cyclic ketoesters and ketolactones with cinchonidine and cinchonine, Synlett 12 (2004) 2115-2118. |
| [7] | À.P. Alexandr Shafir, A. Vallribera, Asymmetric synthesis of L-carbidopa based on a highly enantioselective amination, Org. Lett. 15 (2013) 1448-1451. |
| [8] | T.Y. Liu, H.L. Cui, Y. Zhang, et al., Organocatalytic and highly enantioselective direct amination of aromatic ketones, Org. Lett. 9 (2007) 3671-3674. |
| [9] | T. Bui, G. Hernández-Torres, C. Milite, C.F. Barbas, Highly enantioselective organocatalytic a-amination reactions of aryl oxindoles: developing designer multifunctional alkaloid catalysts, Org. Lett. 12 (2010) 5696-5699. |
| [10] | S. Saaby, M. Bella, Asymmetric construction of quaternary stereocenters by direct organocatalytic amination of α-cyanoacetates and β-dicarbonyl compound, J. Am. Chem. Soc. 126 (2004) 8120-8121. |
| [11] | X. Liu, H. Li, L. Deng, Highly enantioselective amination of α-cyanoacetates with chiral catalysts accessible from both quinine and quinidine, Org. Lett. 7 (2005) 167-169. |
| [12] | K. Juhl, K.A. Jørgensen, Catalytic asymmetric direct α-amination reactions of 2-keto ester: a simple synthetic approach to optically active syn-amino-α-hydroxy esters, J. Am. Chem. Soc. 124 (2002) 2420-2421. |
| [13] | N. Kumaragurubaran, K. Juhl, W. Zhuang, A. Bøgevig, K.A. Jørgensen, Direct L-proline catalyzed asymmetric a-amination of ketone, J. Am. Chem. Soc. 124 (2002) 6254-6255. |
| [14] | B. List, Direct catalytic asymmetric a-amination of aldehydes, J. Am. Chem. Soc. 124 (2002) 5656-5657. |
| [15] | H. Konishi, T.Y. Lam, J.P. Malerich, V.H. Rawal, Enantioselective α-amination of 1,3-dicarbonyl compounds using squaramide derivatives as hydrogen bonding catalysts, Org. Lett. 12 (2010) 2028-2031. |
| [16] | J.Y. Fu, Q.C. Yang, Q.L. Wang, et al., Enantioselective a-amination of branched aldehydes promoted by simple chiral primary amino acids, J. Org. Chem. 76 (2011) 4661-4664. |
| [17] | B. Liu, X. Han, Z. Dong, et al., Highly enantioselective michael addition of 1,3-dicarbonyl compounds to nitroalkenes catalyzed by designer chiral BINOL-quinine-squaramide: efficient access to optically active nitro-alkanes and their isoxazole derivatives, Tetrahedron: Asymmetry 24 (2013) 1276-1280. |
| [18] | Z. Dong, G. Qiu, H.B. Zhou, C. Dong, Chiral squaramide as multiple H-bond donor organocatalysts for the asymmetric Michael addition of 1,3-dicarbonyl compounds to nitroolefins, Tetrahedron: Asymmetry 23 (2012) 1550-1556. |
| [19] | X. Han, B. Liu, H.B. Zhou, C. Dong, Enhanced efficiency of recyclable C3-symmetric cinchonine-squaramides in the asymmetric Friedel-Crafts reaction of indoles with alkyl trifluoropyruvate, Tetrahedron: Asymmetry 23 (2012) 1331-1337. |
| [20] | X. Han, X. Wu, C. Min, H.B. Zhou, C. Dong, An expedient approach to highly enantioenriched cyclic nitrones mediated by robust and recoverable C3-symmetric cinchonine-squaramide catalysts, RSC Adv. 2 (2012) 7501-7505. |
| [21] | X. Han, C. Dong, H.B. Zhou, C3-symmetric cinchonine-squaramide-catalyzed asymmetric chloroactonization of styrene-type carboxylic acids with 1,3-dichloro-5,5-dimethylhydantion: an efficient method to chiral isochroman-1-ones, Adv. Synth. Catal. 356 (2014) 1275-1280. |
| [22] | A.G. Doyle, E.N. Jacobsen, Small molecule H-bond donors in asymmetric catalysis, Chem. Rev. 107 (2007) 5713-5743. |
| [23] | J.D. McGilvra, V.B. Gondi, V.H. Rawal, in: P.I. Dalko (Ed.), Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007, pp. 189-254. |
| [24] | X. Yu, W. Wang, Hydrogen-bond-mediated asymmetric catalysis, Chem. Asian J. 3 (2008) 516-532. |
| [25] | J.P. Malerich, K. Hagihara, V.H. Rawal, Chiral squaramide derivatives are excellent hydrogen bond donor catalysts, J. Am. Chem. Soc. 130 (2008) 14416-14417. |
| [26] | W. Yang, D.M. Du, Highly enantioselective Michael addition of nitroalkanes to chalcones using chiral squaramide as hydrogen bonding organocatalysts, Org. Lett. 12 (2010) 5450-5453. |
| [27] | W. Yang, D.M. Du, Chiral squaramide catalyzed highly enantioselective Michael addition of 2-hydroxy-1,4-naphthoquinones to nitroalkenes, Adv. Synth. Catal. 353 (2011) 1241-1246. |
| [28] | P. Trillo, M.G. Martínez, D.A. Alonso, A. Baeza, 2-Aminobenzimidazole organocatalyzed asymmetric amination of cyclic 1,3-dicarbonyl compounds, Synlett 26 (2015) 95-100. |
| [29] | X. Xu, T. Yabuta, P. Yuan, Y. Takemoto, Organocatalytic enantioselective hydrazination of 1,3-dicarbonyl compounds: asymmetric synthesis of disubstituted a-amino acids, Synlett 1 (2006) 137-140. |
| [30] | K. Hideyuki, T.Y. Lam, J.P. Malerich, V.H. Rawal, Enantioselective α-amination of 1,3-dicarbonyl compounds using squaramide derivatives as hydrogen bonding catalysts, Org. Lett. 12 (2010) 2028-2031. |