




b School of Pharmacy and Department of Chemistry, University of Wisconsin, Madison, WI 53705-2222, USA
Six-membered carbocycles are one of the most ubiquitous ring compounds in organic chemistry. Among the various synthetic routes to build six-membered cycles,transition-metal-catalyzed cycloaddition is widely regarded as one of the most efficient approaches to access highly functionalized carbo- and heterocycles [1]. So far,a number of metal-catalyzed [3 + 3] [2],[4 + 2] [1a, 3], [5 + 1] [4, 5, 6, 7, 8, 9],[2 + 2 + 2] [10],[3 + 2 + 1] [11],and [4 + 1 + 1] [12] reactions have been developed for the synthesis of six-membered carbocycles. However,the formation of a six-membered ring through [5 + 1] cycloaddition has been much less developed, presumably due to the difficulty of searching the appropriate 5- carbon component. The primary 5-carbon components for transition- metal-mediated [5 + 1] cycloadditions have been focused on vinylcyclopropane (VCP) [4] and their related derivatives [5]. Meanwhile,allenylcyclopropanes [6] and cyclopropenes [7] have also been adopted as the 5-carbon synthons in some [5 + 1] cycloadditions. Recently,Malacria and co-workers reported a new class of [5 + 1] cycloadditions using 3-acyloxy-1,4-enyne (ACE) in place of VCP as 5-carbon synthon [8]. This methodology provides an efficient route to the six-membered carbocyclic system of aromatic resorcinols (Scheme 1),a series of valuable compounds with important bioactivity [13] and antibacterial activities [14]. More recently,Tang et al. developed a tandem annulation and [5 + 1] cycloaddition of 1,4-enynes to successfully construct substituted carbazoles,dibenzofurans,and tricyclic compounds containing a cyclohexadienone moiety [15].

|
Download:
|
| Scheme 1.[RhCl(CO)2]2-catalyzed [5 + 1] cycloaddition of ACE and CO. | |
Compared with the experimental discoveries of several modes of [5 + 1] cycloaddition,theoretical studies on the mechanisms of metal-catalyzed [5 + 1] cycloadditions are even rare. Yu group has reported their density functional theory (DFT) calculations on the mechanism of the [5 + 1] cycloaddition of 1-yne-vinylcyclopropanes and two CO units [5c]. To our best knowledge,the mechanism of the [5 + 1] cycloaddition of ACE and CO remains unknown till now. The previous experimental studies by Malacria [8] suggested several pathways for the formation of the resorcinols through [5 + 1] cycloaddition of ACE and CO (Scheme S1 in Supporting information). It is not clear which pathway is actually preferred and which step determines the reaction rate. To answer these questions,we performed theoretical computations on these pathways. In addition,the effect of ester on the reactivity of this [5 + 1] cycloaddition is also experimentally found to be interesting [16]. As shown in Table 1,significant enhancement of reactivitywas observed for benzoate substrate bearing an electron-donating dimethylamino group. For examples,p-dimethylaminobenzoate 1c can react with CO at 50 ℃ to generate the desired product in good yield in 1 h.While pivalate 1b does not work at the same condition but requires higher CO pressure,higher temperature and longer reaction time (entry 3 vs. entries 2 and 2'). Significantly,p-dimethylaminobenzoate with methyl substituent at C3 (substrate 1f) can react with CO at room temperature (entry 6). In contrast, substrate 1e does not work at room temperature (entry 5). Clearly, the electron-rich p-dimethylaminobenzoate dramatically increases the reactivity. In this work,computational investigations on [RhCl(CO)2]2-catalyzed intermolecular [5 + 1] cycloadditions between 3-acyloxy-1,4-enynes (ACEs) and CO have disclosed the preferred mechanism and origins of electronic effects of substrates on reactivities. These results may have broad implications in understanding and design of transition-metal-catalyzed reactions.
|
|
Table 1 Experimental results and computed activation free energies for [5 + 1] cycloadditions of ACE derivatives and CO. |

{kind=link}
Geometry optimization and frequency analysis were performed in CH2Cl2 solvent with the SMD solvation model using B3LYP and a mixed basis set of SDD for Rh and 6-31G(d) for other atoms. Single point energies were calculated at the M06/6-311+G(d,p) level (SDD for Rh) with the SMD model (CH2Cl2 solvent) on B3LYP-optimized geometries. This is the same methodology that we used in our previous theoretical studies of [RhCl(CO)2]2-catalyzed [5 + 2] cycloadditions involving 3-acyloxy-1,4-enynes (ACEs) [17]. All calculations were carried out with Gaussian 09 [18]. All reported free energies involve thermal corrections to Gibbs free energy at 298 K. Computed structures are illustrated using CYLVIEW drawings [19].
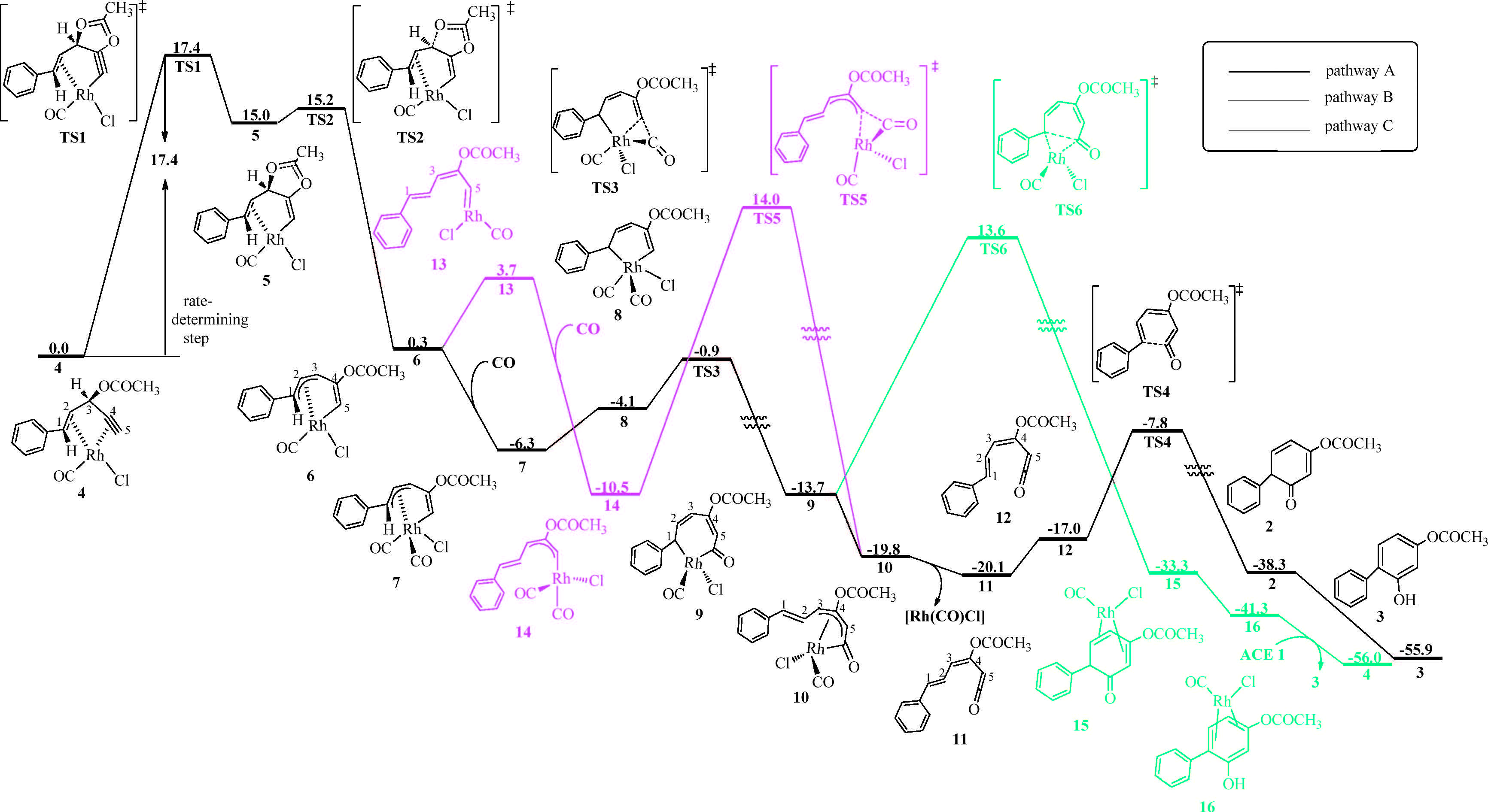
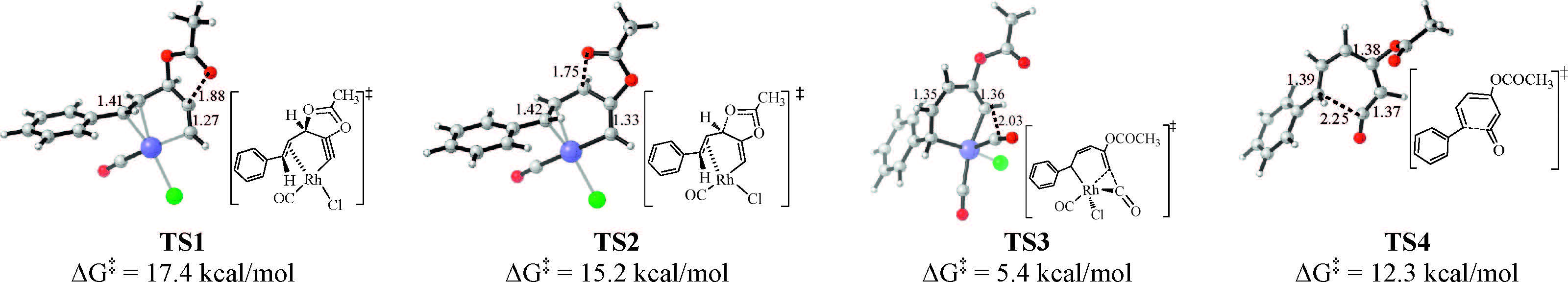
3. Results and discussionWe employed ACE 1 as the model substrate and computed the mechanism of the [5 + 1] cycloaddition of ACE 1 and CO. The computed Gibbs free energy profiles of several possible reaction pathways are shown in Fig. 1. Initially,the rhodium dimer [RhCl(CO)2]2 dissociates [20] and coordinates to the ACE,leading to the formation of a ACE-Rh(CO)Cl π complex 4 [21]. This complex enters the catalytic cycle. Among the three possible pathways A,B and C,pathway A (in black line) is calculated to be the most favored mechanism. Along pathway A,firstly,the stepwise 1,2-acyloxy migration occurs via intermediate 5 to afford 6. In this step Rh(I) is oxidized to Rh(II). Then coordination of CO to rhodium center generates 7. Subsequently, CO insertion into the Rh-C(sp2) bond affords the sevenmembered metallacycle 9,which then isomerizes to a more stable species 10 by rupture of Rh-C1 bond followed by rotation of the C2-C3 bond,leading to the styryl group positioned far from the rhodium center in 10. Then reductive elimination of 10 affords the ketene 11 and the simultaneous release of the active catalyst Rh(CO)Cl,which coordinates to another ACE 1 molecule to start the next catalytic cycle. Isomerization of 11 may afford 12 by rotating the C2-C3 bond. Next,6π-electroncyclization of 12 leads to product 2,which can undergo aromatization to afford the resorcinol product 3. In pathway A,the ratedetermining step is 1,2-acyloxy migration,requiring an activation free energy of 17.4 kcal/mol (TS1,Fig. 1). The energy barrier for CO insertion is 5.4 kcal/mol (from 7 to TS3). Reductive elimination of 10 and the subsequent 6pelectroncyclization of 11 needs to overcome an overall free energy barrier of 12.3 kcal/mol (from 11 to TS4). The geometries of the key transition states involved in this favored pathway are shown in Fig. 2.

|
Download:
|
| Fig. 1.Gibbs free energy profile of the [RhCl(CO)2]2-catalyzed intermolecular [5 + 1] cycloaddition of ACE 1 and CO. Energies are in kcal/mol and calculated using M06/SDD-6-311+G(d,p)/SMD(CH2Cl2)//B3LYP/SDD-6-31G(d)/SMD(CH2Cl2). | |
{kind=link}

|
Download:
|
| Fig. 2.Transition states in pathway A of the [RhCl(CO)2]2-catalyzed intermolecular [5 + 1] cycloaddition of ACE 1 and CO. | |
{kind=link}
In pathway A,the intermediate 6 isomerizes to carbene 13,in which the C1 atom is positioned far away from the rhodium center. Coordination of CO to rhodium center then forms intermediate 14. Subsequently,CO insertion into the Rh-C(sp2) bond (TS5) generates intermediate 10. This step requires an activation free energy of 24.5 kcal/mol. The overall activation free energy (from 14 to TS5) of pathway B is higher than that of the rate-determining step of pathway A. The high energy of TS5 may be attributed to the unstable 12-electron structure of Rh in TS5. Therefore,we considered another possible transition state TS5-1 that possesses two CO ligands. The calculation results showed that the energy of TS5-1 is also higher than that of TS3 (Fig S2 in Supporting information). Thus,pathway B should be ruled out from the favored pathways.
In pathway C,reductive elimination of 9 affords the complex 15. Aromatization of 15 leads to resorcinol complex 16,which undergoes ligand exchange to produce resorcinol product 3. This pathway requires an activation free energy of 27.3 kcal/mol (from 9 to TS6),much higher than that of the rate-determining step of pathway A. Because the formation of a new bond between two double bonds in TS4 is easier than that between two single bonds in TS6,TS4 is lower in energy than TS6. Therefore,pathway C should be ruled out from the favored pathways too. Based on the above data,pathway A is proposed as the most preferred mechanism.
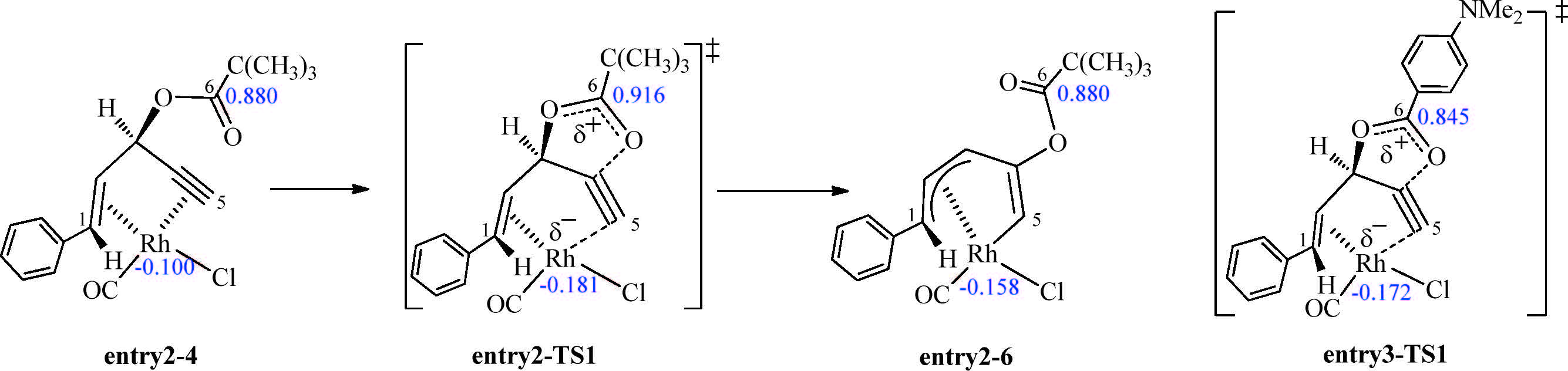
We then investigated the effects of substituents on the reactivity of the reaction,which is determined by the barrier of 1,2-acyloxy migration. As shown in Table 1,experimentally, electron-rich esters dramatically increase the reactivity of ACEs in the [5 + 1] cycloaddition [16]. This is similar to the Rh-catalyzed intermolecular [5 + 2] cycloadditions of ACEs and alkynes [22]. The computed free energy barriers of the 1,2-acyloxy migration steps provide good agreement with the experimental reactivities of different esters (Table 1). This is because that the electrondonating group stabilizes the positive charge building up in the oxocyclic transition state as illustrated in Fig. 3. Moreover, computational results show that electronic effects on the rate of the alkyne insertion,the reductive elimination,and 6π-electroncyclization steps are very small (Table S1 in Supporting information). These results confirm that 1,2-acyloxy migration is the rate-determining step and the different reactivities of ACE substrates are due to the effects on 1,2-acyloxy migration. The activation barriers for acyloxy migration of electron-deficient ester 1 g (R1 = -CF3,entry 7) increases dramatically compared with those of other esters listed in Table 1. The activation barrier for acyloxy migration of p-nitrobenzoate which bears an electronwithdrawing NO2 group (entry 8) also greatly increases compared with that of the electron-rich p-dimethylaminobenzoate (entry 3). These results indicate that electron-deficient esters should be unfavorable for this reaction. The computed barrier of ACE 1i (R2 = Me,entry 9) is slightly increased compared with that of ACE 1c (R2 = Ph,entry 3) due to weaker electron-donating ability of methyl group than phenyl group. Significantly,because the substituent at C3 stabilizes the allyl intermediate via hyperconjugation and lowers the barrier of acyloxy migration,the ACE with alkyl substitution at C3 requires relative lower 1,2-acyloxy migration barrier than ACE without alkyl substitution (Table 1). As a result,the reaction of substrate 1f occurs at room temperature, while that of 1c takes place at 50 ℃ (entries 6 and 3 in Table 1). The pivalate 1e is predicted to react at 50 ℃ or at higher temperature according to the calculated 1,2-acyloxy migration barrier although it does not react at room temperature (entry 5). The computed barrier of ACE 1j (R3 = Et,entry 10) is slightly decreased compared with that of ACE 1f (R3 = Me,entry 6) due to stronger hyperconjugation effect of ethyl group than methyl group. Moreover,the above computational results imply that the reactivities should be determined by different ester substrates and be irrelevant to CO for the [5 + 1] cycloaddition or irrelevant to alkynes for the [5 + 2] cycloaddition,since only ACE is involved in the rate-determining acyloxy migration for both [5 + 1] and [5 + 2] [17] cycloadditions. This is consistent with experiments,for example,both [5 + 1] and [5 + 2] reactions with substrate 1f take place at room temperature.

|
Download:
|
| Fig. 3.NPA charges of transition states entry2-TS1 and entry3-TS1 in Table 1. In the rate-determining 1,2-acyloxy migration step, the ester carbon (C6) is more positively charged in the transition state (TS1) than in the reactant (4) and the product (6). Thus, the electron-rich p-dimethylaminobenzoate substrate stabilizes the positive charge on C6 in the acyloxy migration transition state (entry3-TS1) and promotes the reaction. As expected, the C6 atom in entry3-TS1 is less positively charged than that in the transition state with the pivalate substrate (entry2-TS1). | |
{kind=link}
In summary,we performed DFT calculations to explore the mechanism of [RhCl(CO)2]2-catalyzed [5 + 1] cycloaddition of ACE and CO and disclosed the origins of substituent effect on reactivity. The catalytic cycle involves a stepwise 1,2-acyloxy migration to form a rhodium-allyl intermediate,CO insertion into the Rh-C(sp2) bond to generate a seven-membered metallacycle intermediate, reductive elimination to afford ketene,6π-electroncyclization and subsequent aromatization to produce the resorcinol product. The 1,2-acyloxy migration is predicted to be the rate-determining step, which is consistent with higher reactivity experimentally observed for ACEs bearing an electron-rich benzoate. Our theoretical studies disclosed here should have broad implications in understanding transition-metal-catalyzed cycloadditions and may facilitate the design of new types of reactions.
AcknowledgmentsWe are grateful to Tianjin Natural Science Foundation (No. 14JCYBJC20100 X.X.),andMOE Innovation Teams (Nos. IRT-13R30 and IRT13022) of China,and NIH (No. R01GM088285 W.T.) for financial support. Calculations were performed on the TH-1A cluster at National Supercomputer Center in Tianjin.
Appendix A. Supplementary dataSupplementary material related to this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.03.016.
| [1] | For selected examples, see: (a) M Lautens, W. Klute, W. Tam, Transition metal-mediated cycloaddition reactions, Chem. Rev. 96 (1996) 49-92;(b) H.W. Fruhauf, Metal-assisted cycloaddition reactions in organotransition metal chemistry, Chem. Rev. 97 (1997) 523-596; (c) B.M. Trost, M.J. Krische, Transition metal catalyzed cycloisomerizations, Synlett (1998) 1-16; (d) C. Aubert, O. Buisine, M. Malacria, The behavior of 1,n-enynes in the presence of transition metals, Chem. Rev. 102 (2002) 813-834; (e) P.A. Evans, Modern Rhodium-Catalyzed Organic Reactions, Wiley-VCH, Weinheim, 2005; (f) V. Michelet, P.Y. Toullec, J.P. Genet, Cycloisomerization of 1,n-enynes: challenging metal-catalyzed rearrangements and mechanistic insights, Angew. Chem. Int. Ed. 47 (2008) 4268-4315; (g) Z.X. Yu, Y. Wang, Y. Wang, Transition-metal-catalyzed cycloadditions for the synthesis of eight-membered carbocycles, Chem. Asian J. 5 (2010) 1072-1088; (h) P.A. Inglesby, P.A. Evans, Stereoselective transition metal-catalysed higherorder carbocyclisation reactions, Chem. Soc. Rev. 39 (2010) 2791-2805; (i) Z. Chen, X. Han, J.H. Liang, et al., Cycloaddition reactions of benzyne with olefins, Chin. Chem. Lett. 25 (2014) 1535-1539; (j) H. Mehrabi, M. Hatami-Pour, Facile, one-pot synthesis of new phenanthridine derivatives through 1,4-dipolar cycloaddition of phenanthridine, activated acetylenes, and aromatic aldehydes, Chin. Chem. Lett. 25 (2014) 1495-1498. |
| [2] | (a) Y. Huang, X. Lu, Palladium catalyzed annulation reaction using a bifunctional allylic alkylating agent, Tetrahedron Lett. 29 (1988) 5663-5664; (b) X.C. He, B. Wang, B.D. Bai, Studies on asymmetric synthesis of huperzine A-1. Palladium-catalyzed asymmetric bicycloannulation of 5,6,7,8-tetrahydro-2-methoxy-6-oxo-5-quinolinecarboxylic esters, Tetrahedron Lett. 39 (1998) 411-414. |
| [3] | (a) W. Carruthers, Cycloaddition Reactions in Organic Synthesis, Pergamon, Oxford, 1990; (b) P.A. Wender, T.E. Jenkins, S. Suzuki, Transition metal-catalyzed intramolecular[4 + 2] Diene-Allene cycloadditions: a convenient synthesis of angularly substituted ring systems with provision for catalyst-controlled chemo-and stereo-complementarity, J. Am. Chem. Soc. 117 (1995) 1843-1844; (c) D.J.R. O'Mahony, D.B. Belanger, T. Livinghouse, Substrate control of stereoselection in the rhodium(I) catalyzed intramolecular [4 + 2] cycloaddition reaction, Org. Biomol. Chem. 1 (2003) 2038-2040; (d) K. Aikawa, S. Akutagawa, K. Mikami, Asymmetric synergy between chiral dienes and diphosphines in cationic Rh(I)-catalyzed intramolecular [4 + 2] cycloaddition, J. Am. Chem. Soc. 128 (2006) 12648-12649; (e) A. Fürstner, C.C. Stimson, Two manifolds for metal-catalyzed intramolecular Diels-Alder reactions of unactivated alkynes, Angew. Chem. Int. Ed. 46 (2007) 8845-8849; (f) H. Kusama, Y. Karibe, Y. Onizawa, N. Iwasawa, Gold-catalyzed tandem cyclization of dienol silyl ethers for the preparation of bicyclo[4.3.0]nonane derivatives, Angew. Chem. Int. Ed. 49 (2010) 4269-4272; (g) S.M. Kim, J.H. Park, Y.K. Chung, Au(PPh3)OPOF2-catalyzed intramolecular[4 + 2] cycloaddition reaction of dienynes, Chem. Commun. 47 (2011) 6719-6721. |
| [4] | (a) R. Aumann, Reactions of strained carbon-carbon bonds with transition metals. 7. Iron carbonyl complexes from vinylcyclopropane, J. Am. Chem. Soc. 96 (1974) 2631-2632; (b) D.F. Taber, K. Kanai, Q. Jiang, G. Bui, Enantiomerically pure cyclohexenones by Fe-mediated carbonylation of alkenyl cyclopropanes, J. Am. Chem. Soc. 122 (2000) 6807-6808; (c) D.F. Taber, P.V. Joshi, K. Kanai, 2,5-Dialkyl cyclohexenones by Fe(CO)5-mediated carbonylation of alkenyl cyclopropanes: functional group compatibility, J. Org. Chem. 69 (2004) 2268-2271; (d) T. Kurahashi, A. de Meijere, Cyclopropyl building blocks for organic synthesis, Part 120. [5 + 1] cocyclization of (cyclopropylmethylene)cyclopropanes and other vinyl-cyclopropanes with carbon monoxide catalyzed by octacarbonyldicobalt, Synlett (2005) 2619-2622. |
| [5] | (a) P.A. Wender, G.G. Gamber, R.D. Hubbard, S.M. Pham, L. Zhang, Multicomponent cycloadditions: the four-component [5 + 1 + 2 + 1] cycloaddition of vinylcyclopropanes, alkynes, and CO, J. Am. Chem. Soc. 127 (2005) 2836-2837; (b) Z.K. Yao, J. Li, Z.X. Yu, Rh-catalyzed [7 + 1] cycloaddition of buta-1,3-dienylcyclopropanes and CO for the synthesis of cyclooctadienones, Org. Lett. 13 (2011) 134-137; (c) M. Lin, F. Li, L. Jiao, Z.X. Yu, Rh(I)-catalyzed formal [5 + 1]/[2 + 2 + 1] cycloaddition of 1-yne-vinylcyclopropanes and two CO units: one-step construction of multifunctional angular tricyclic 5/5/6 compounds, J. Am. Chem. Soc. 133 (2011) 1690-1693. |
| [6] | (a) N. Iwasawa, Y. Owada, T. Matsuo, Octacarbonyldicobalt promoted transformation of 1-(1,2-propadienyl) cyclopropanols to 1,4-hydroquinones, Chem. Lett. (1995) 115-116; (b) Y. Owada, T. Matsuo, N. Iwasawa, Transformation of 1-(1,2-propadienyl) cyclopropanols into substituted hydroquinones employing octacarbonyldicobalt, Tetrahedron 53 (1997) 11069-11086; (c) M. Murakami, K. Itami, M. Ubukata, I. Tsuji, Y. Ito, Iridium-catalyzed [5 + 1] cycloaddition: allenylcyclopropane as a five-carbon assembling unit, J. Org. Chem. 63 (1998) 4-5; (d) D. Shu, X. Li, M. Zhang, P.J. Robichaux, W. Tang, Synthesis of highly functionalized cyclohexenone rings: rhodium-catalyzed 1,3-acyloxy migration and subsequent[5 + 1] cycloaddition, Angew. Chem. Int. Ed. 50 (2011) 1346-1349; (e) D. Shu, X. Li, M. Zhang, P.J. Robichaux, I.A. Guzei, W. Tang, Rhodium-catalyzed carbonylation of cyclopropyl substituted propargyl esters: a tandem 1,3-acyloxy migration [5 + 1] cycloaddition, J. Org. Chem. 77 (2012) 6463-6472. |
| [7] | (a) N.A. Grabowski, R.P. Hughes, B.S. Jaynes, A.L. Rheingold, Stepwise transition metal promoted ring expansion reactions of vinylcyclopropenes to give cyclopentadienes and cyclohexa-2,4-dienones. The first example of a 1-metallacyclohexa-2,4-diene complex, {[Pt-CH2-CH≡C(Ph)-C(Ph)≡C(Ph)](PPh3)2}, J. Chem. Soc. Chem. Commun. (1986) 1694-1695; (b) S.H. Cho, L.S. Liebeskind, Practical organic synthesis with strained ring molecules. Rhodium catalyzed carbonylation of cyclopropenecarboxylate esters and cyclopropenyl ketones to a-pyrones and/of vinyl cyclopropenes to phenols, J. Org. Chem. 52 (1987) 2631-2634; (c) M.F. Semmelhack, S. Ho, M. Steigerwald, M.C. Lee, Metal carbonyl-promoted rearrangement of cyclopropenes to naphthols, J. Am. Chem. Soc. 109 (1987) 4397-4399; (d) M.F. Semmelhack, S. Ho, D. Cohen, et al., Metal-catalyzed cyclopropene rearrangements for benzannulation: evaluation of an anthraquinone synthesis pathway and reevaluation of the parallel approach via carbene-chromium complexes, J. Am. Chem. Soc. 116 (1994) 7108-7122. |
| [8] | (a) C. Brancour, T. Fukuyama, Y. Ohta, et al., Synthesis of functionalized resorcinols by rhodium-catalyzed [5 + 1] cycloaddition reaction of 3-acyloxy-1,4-enynes with CO, Chem. Commun. 46 (2010) 5470-5472; (b) T. Fukuyama, Y. Ohta, C. Brancour, et al., Rh-catalyzed [5 + 1] and [4 + 1] cycloaddition reactions of 1,4-enyne esters with CO: a shortcut to functionalized resorcinols and cyclopentenones, Chem. Eur. J. 18 (2012) 7243-7247. |
| [9] | A. Kamitani, N. Chatani, T. Morimoto, S. Murai, Carbonylative [5 + 1] cycloaddition of cyclopropyl imines catalyzed by ruthenium carbonyl complex, J. Org. Chem. 65 (2000) 9230-9233. |
| [10] | (a) S. Kotha, E. Brahmachary, K. Lahiri, Transition metal catalyzed [2 + 2 + 2] cycloaddition and application in organic synthesis, Eur. J. Org. Chem. (2005) 4741-4767; (b) V. Gandon, C. Aubert, M. Malacria, Recent progress in cobalt-mediated[2 + 2 + 2] cycloaddition reactions, Chem. Commun. (2006) 2209-2217; (c) P.R. Chopade, J. Louie, [2 + 2 + 2] cycloaddition reactions catalyzed by transition metal complexes, Adv. Synth. Catal. 348 (2006) 2307-2327; (d) W. Wu, X.Y. Zhang, S.H. Kang, Rhodium-catalyzed selective [2 + 2 + 2] cyclizations of 1,6-diyneswithmonoynes leading to isoindolines and isobenzofurans, Chin. Chem. Lett. 21 (2010) 18-22. |
| [11] | (a) Y. Koga, K. Narasaka, Rhodium catalyzed transformation of 4-pentynyl cyclopropanes to bicyclo[4.3.0]nonenones via cleavage of cyclopropane ring, Chem.Lett. (1999) 705-706; (b) S.I. Lee, J.H. Park, Y.K. Chung, S.G. Lee, Rhodium-catalyzed carbonylative[3 + 2 + 1] cycloaddition reaction: catalytic formation of bicyclic cyclohexenones from trienes and carbon monoxide, J. Am. Chem. Soc. 126 (2004) 2714-2715; (c) L. Jiao, M. Lin, L.G. Zhuo, Z.X. Yu, Rh(I)-catalyzed [(3 + 2) + 1] cycloaddition of 1-yne/ene-vinylcyclopropanes and CO: homologous Pauson-Khand reaction and total synthesis of (±)-α-agarofuran, Org. Lett. 12 (2010) 2528-2531; (d) C. Li, H. Zhang, J. Feng, Y. Zhang, J. Wang, Rh(I)-catalyzed carbonylative carbocyclization of tethered ene-and yne-cyclopropenes, Org. Lett. 12 (2010) 3082-3085. |
| [12] | D.F. Taber, P. Guo, N. Guo, Intramolecular [1 + 4 + 1] cycloaddition: establishment of the method, J. Am. Chem. Soc. 132 (2010) 11179-11182. |
| [13] | A. Kozubek, J.H.P. Tyman, Resorcinolic lipids, the natural non-isoprenoid phenolic amphiphiles and their biological activity, Chem. Rev. 99 (1999) 1-26. |
| [14] | M. Himejima, I. Kubo, Antibacterial agents from the cashew anacardium occidentale (anacardiaceae) nut shell oil, J. Agric. Food Chem. 39 (1991) 418-421. |
| [15] | X. Li, W. Song, W. Tang, Rhodium-catalyzed tandem annulation and (5 + 1) cycloaddition: 3-hydroxy-1,4-enyne as the 5-carbon component, J. Am. Chem. Soc. 135 (2013) 16797-16800. |
| [16] | C.M. Schienebeck, W. Song, A.M. Smits, W. Tang, Rhodium-catalyzed intermolecular[5 + 1] and [5 + 2] cycloadditions using 1,4-enynes with an electron-donating ester on the 3-position, Synthesis (2015), http://dx.doi.org/10.1055/s-0034-1380160. |
| [17] | X. Xu, P. Liu, X.Z. Shu, W. Tang, K.N. Houk, Rh-catalyzed (5 + 2) cycloadditions of 3-acyloxy-1,4-enynes and alkynes: computational study of mechanism, reactivity, and regioselectivity, J. Am. Chem. Soc. 135 (2013) 9271-9274. |
| [18] | M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010. |
| [19] | C.Y. Legault, CYLview, 1.0b, Universitéde Sherbrooke, Sherbrooke, Québec, Canada, 2009, http://www.cylview.org. |
| [20] | M.R. Wilson, A. Prock, W.P. Giering, p Effects involving Rh-PZ3 compounds. The quantitative analysis of ligand effects (QALE), Organometallics 21 (2002) 2758-2763. |
| [21] | Computational studies involving the active catalyst Rh(CO)Cl which derives from dissociation of the dimer [RhCl(CO)2]2: (a) ZX. Yu, P.A. Wender, K.N. Houk, On the mechanism of [Rh(CO)2Cl]2-catalyzed intermolecular (5 + 2) reactions between vinylcyclopropanes and alkynes, J. Am. Chem. Soc. 126 (2004) 9154-9155; (b) Z.X. Yu, P.H.Y. Cheong, P. Liu, et al., Origins of differences in reactivities of alkenes, alkynes, and allenes in [Rh(CO)2Cl]2-catalyzed (5 + 2) cycloaddition reactions with vinylcyclopropanes, J. Am. Chem. Soc. 130 (2008) 2378-2379; (c) P. Liu, P.H.Y. Cheong, Z.X. Yu, P.A. Wender, K.N. Houk, Substituent effects, reactant preorganization, and ligand exchange control the reactivity in Rh(I)-catalyzed (5 + 2) cycloadditions between vinylcyclopropanes and alkynes, Angew. Chem. Int. Ed. 47 (2008) 3939-3941; (d) X. Xu, P. Liu, A. Lesser, et al., Ligand effects on rates and regioselectivities of Rh(I)-catalyzed (5 + 2) cycloadditions: a computational study of cyclooctadiene and dinaphthocyclooctatetraene as ligands, J. Am. Chem. Soc. 134 (2012) 11012-11025. |
| [22] | C.M. Schienebeck, P.J. Robichaux, X. Li, L. Chen, W. Tang, Effect of ester on rhodium-catalyzed intermolecular [5 + 2] cycloaddition of 3-acyloxy-1,4-enynes and alkynes, Chem. Commun. 49 (2013) 2616-2618. |