




b Chemistry Department, Faculty of Science (Girl's), Al-Azhar University, Nasr City, Cairo, Egypt;
c Chemistry Department, Faculty of Science, Al-Azhar University, Nasr City, Cairo, Egypt;
d Photochemistry Department, National Research Center, El-Dokki, Giza, Egypt
The pyrimidine moiety is one of the most widespread heterocycles frombiologically occurring compounds such as nucleic acids and vitamin B1,and is an important constituent of numerous drug molecules in many therapeutic areas [1, 2, 3]. Aminopyrimidines are common in marketing drugs such as anti-atherosclerotic aronixil, anti-histaminic thonzylamine,anti-anxielytic buspirone,anti-psoriatic enazadrem and other medicinally relevant compound (Fig. 1) [4]. Guanidines are important applications in many fields,such as pharmaceutics,organometallic and coordination chemistry,and organic synthesis [5, 6, 7, 8]. Various procedures for the synthesis of amino-pyrimidine derivatives have been described. For example, the reaction of guanidine derivatives with a variety of 1,3- dielectrophilic three-carbon units as a,b-unsaturated carbonyl compounds gave aminopyrimidines [9, 10, 11]. Singh and co-workers [12] have reported the synthesis of a series of tertahydroquinazolin- 2-ylamines and cycloheptopyrimidin-2-ylamines by the reaction of bis-benzylidine cycloalkanones and guanidine hydrochloride in the presence of sodium hydride. Multicomponent reactions (MCRs) have emerged as an extremely powerful tool in combinatorial chemistry and drug discovery,since it offers significant advantages over conventional linear syntheses and its termstoimprove classical organic reactions promote new reactions and develop straightforward synthetic routes for bioactive heterocycles [13, 14, 15]andnatural products [16]. In view of the above mentioned benefits and in continuation of our studies [17, 18, 19],we report here the synthesis of some novel cyclopenta[d]pyrimidines 3a-c in excellent yields via the one-pot multicomponenet reactions of cyclopentanone 1, aromatic aldehyde and guanidine hydrochloride. Also,the pyrimido[ 1,2-a]-pyrimidines 5,7and 12 were obtained by cyclization of compound 3c with some electrophilic reagents.

|
Download:
|
| Fig. 1.Biologically active aminopyrimidine derivatives. | |
{kind=link}
Melting points were determined on a digital Gallen-Kamp MFB-595 instrument and uncorrected. IR spectra (KBr) were measured on a Shimadzu 440 spectrometer. The 1H NMR and 13C NMR (DMSO-d6) spectra were recorded with a Varian Gemini 300 (300 MHz) and Varian Gemini 75 MHz spectrometer,respectively.The chemical shift values are expressed in ppm (δ scale) using tetramethylsilane as an internal standard. Mass spectra were performed on a Shimadzu GSMS-QP 1000 Ex mass spectrometer at 70 eV. The elemental analyses were carried out at the Microanalytical Center,Cairo University,Cairo (Egypt).
2.1. General procedure for preparation of 3a-cMethod (A): A mixture of cyclopentanone 1 (0.01 mmol), aromatic aldehyde (0.02 mmol) and guanidine hydrochloride (0.01 mmol) was refluxed in sodium methoxide (0.01 mmol Na in 30 mL methanol) for 3 h. The product was precipitated on heating then filtered off,washed with water,dried,and recrystallized to give 3.
Method (B): A mixture of 2,5-bis(arylmethylidene)cyclopentanone 2 (0.01 mmol),guanidine hydrochloride (0.01 mmol) and sodium methoxide (0.01 mmol Na in 30 mL methanol) was refluxed for 3 h. The resulting solid product was collected by filtration and recrystallized to give 3.
2-Amino-7-(2-hydroxybenzylidene)-4-(2-hydroxyphenyl)- 4,5,6,7-tetrahydro-3H-cyclopenta[d]pyrimidine 3a: This compound was obtained in 79% (2.63 mg) as a brown crystals (from benzene),m.p. 118-119 ℃. IR (KBr,cm-1) νmax 3365,3204 (OH/ NH2),2935,2851 (CH-aliph),1632 (C=N). 1H NMR (DMSO-d6): δ 1.79 (m,2H,CH2),2.90 (m,2H,CH2),3.6 (b,2H,NH2,exchangeable with D2O),5.20 (s,1H,pyrimidine-H),7.21-7.70 (m,10H,Ar-H, NH,=CH),7.93,8.73 (2s,2H,2OH,exchangeable with D2O. 13C NMR (75 MHz,DMSO-d6): δ 23.7,28.3,54.1,116.2,117.2,119.4, 121.5,121.8,123.6,125.2,128.1,128.4,129.2,129.8,133.2,136.4, 140.3,154.6,156.2,161.5. Anal. Calcd. for C20H19N3O2: C,72.05; H, 5.74; N,12.60.Found C,72.00; H,5.60; N,12.60.
2-Amino-7-(4-methoxybenzylidene)-4-(4-methoxyphenyl)- 4,5,6,7-tetrahydro-3H-cyclopenta[d]pyrimidine 3b: This compound was obtained in 85% (3.07 mg) as a pale brown crystals (from ethanol),m.p. 136-137 ℃. IR (KBr,cm-1) νmax 3400,3228 (NH2),2926,2851 (CH-aliph),1614 (C=N). 1H NMR (DMSO-d6): δ 2.17 (m,2H,CH2),2.67 (m,2H,CH2),3.40 (b,2H,NH2,exchangeable with D2O),3.72,3.83 (2s,6H,2OCH3),5.30 (s,1H,pyrimidine-H), 6.45 (s,1H,NH,exchangeable with D2O),6.82-7.75 (m,9H,Ar-H, 55CH); 13C NMR (75 MHz,DMSO-d6):δ23.6,27.3,57.3,58.2,61.4, 114.6,115.4,124.9,126.2,128.6,130.2,132.8,135.4,136.7,141.2, 154.8,159.4,160.5. Anal. Calcd. for C22H23N3O2: C,73.11; H,6.41; N,11.63. Found: C,73.00; H,6.38; N,11.60.
2-Amino-7-(4-chlorobenzylidene)-4-(4-chlorophenyl)-4,5,6,7- tetrahydro-3H-cyclopenta[d]pyrimidine 3c: This compound was obtained in 76% (2.81 mg) as a brown crystals (from benzene),m.p. 120-121 ℃. IR (KBr,cm-1) νmax 3364,3300 (NH2),2954,2820 (CH-aliph),1624 (C=N). 1H NMR (DMSO-d6) δ: 2.16 (m,2H,CH2), 2.79 (m,2H,CH2),3.15 (b,2H,NH2,exchangeable with D2O),5.30 (s,1H,pyrimidine-H),7.09-7.50 (m,9H,Ar-H,55CH),7.80 (s,1H, NH,exchangeable with D2O); 13C NMR (75 MHz,DMSO-d6): δ 23.6, 27.1,60.6,125.4,127.3,128.5,128.8,129.4,132.7,133.0,133.4, 133.9,136.1,139.9,141.2,154.2; MS (m/z,%): 369 (M+,42.9),370 (M + 1,22.4),371 (M + 2,9.5),368 (M-1,98.6),366 (base peak),329 (29),281 (20),244 (17),206 (68),150 (27),115 (70),76 (8.7); Anal. Calcd. for C20H17Cl2N3: C,64.87; H,4.63; N,11.35.Found C,64.90; H,4.60; N,11.30.
2.2. 4-Amino-9-(4-chlorobenzylidene)-6-(4-chlorophenyl)- 2-(methylthio)-6,7,8,9-tetrahydro-cyclopenta[d]pyrimido [1,2-a]pyrimidine-3-carbonitrile 5A mixture of compound 3c (0.01 mmol) and bis(methylthio)- methylene malononitrile 4 (0.01 mmol) in N,N-dimethylformamide (10 mL) and anhydrous potassium carbonate (15 mg) was refluxed for 3 h. Then,the reaction mixture was poured into ice water. The solid product that formed was filtered off,dried and recrystallized to give 5. This compound was obtained in 75% (3.69 mg) as a dark brown crystals (from ethanol),m.p. 250- 251 ℃. IR (KBr,cm-1) νmax 3319,3196 (NH2),2923,2896 (CH- aliph),2207 (C≡N). 1H NMR (DMSO-d6): δ 2.20 (m,2H,CH2),2.49 (s,3H,SCH3),2.90 (m,2H,CH2),5.20 (s,1H,pyrimidine-H),6.80- 7.60 (m,9H,Ar-H,55CH),8.82 (b,2H,NH2,exchangeable with D2O); 13C NMR (75 MHz,DMSO-d6): δ 19.3,23.2,28.1,58.2,88.7, 115.4,125.5,126.4,128.4,128.9,129.7,132.4,132.9,134.2,136.0, 140.2,141.8,154.4,160.30,169.0; Anal. Calcd. for C25H19Cl2N5S: C, 60.97; H,3.89; N,14.22.Found C,60.90; H,3.80; N,14.20.
2.3. 4-Amino-9-(4-chlorobenzylidene)-6-(4-chlorophenyl)-2- (3,4,5-trimethoxyphenyl)-6,7,8,9-tetrahydrocyclopenta[ d]pyrimido[1,2-a]pyrimidine-3-carbonitrile 7A mixture of compound 3c (0.01 mmol),3,4,5-trimethoxybenzylidenemalononitrile 6 (0.01 mmol) and piperidine (0.1 mL) in dioxane (30 mL) was refluxed for 3 h. The reaction mixture was cooled,then poured onto ice and acidified with hydrochloric acid. The solid product that formed was filtered off,dried and recrystallized to give 7. This compound was obtained in 81% (49.61 mg) as a greenish crystals (from ethanol),m.p. 180-181 ℃ IR (KBr,cm-1) νmax 3350,3235 (NH2),2932 (CH-aliph),2190 (CBBN). 1H NMR (DMSO-d6) δ: 1.85 (m,2H,CH2),2.90 (m,2H,CH2), 3.7 (s,3H,OCH3),3.83 (s,6H,2OCH3),5.2 (s,1H,pyrimidine-H), 6.70 (s,2H,NH2,exchangeable with D2O),7.38-7.60 (m,11H,Ar-H, 55CH); 13C NMR (75 MHz,DMSO-d6) δ: 23.6,28.4,56.6,58.7,61.4, 68.2,104.7,116.4,125.3,126.7,128.7,128.9,129.2,129.8,132.4, 132.9,134.4,135.3,138.7,140.2,141.8,153.1,153.8,160.2,167.1;MS (m/z,%): 612 (M + 1,12.6),613 (M + 2,9.9),552 (11.7),492 (19.8),444 (20.4),405 (11.1),370 (27.8),325 (14),278 (14.2),245 (92),181 (40),125 (base peak),119 (80),90 (4). Anal. Calcd. for C33H27Cl2N5O3: C,64.70; H,4.44; N,11.43.Found C,64.40; H,4.50; N,11.20.
2.4. Ethyl3-((7-(4-chlorobenzylidene)-4-(4-chlorophenyl)-4,5,6,7- tetrahydro-3H-cyclopenta-[d]pyrimidin-2-yl)amino)-2- cyanoacrylate 9A mixture of compound 3c (0.01 mmol),ethoxymethylenecyanoacetate 8 (0.01 mmol) and piperidine (0.1 mL) in ethanol (30 mL) was refluxed for 3 h. The reaction mixture was cooled, then the resulting solid product was collected by filtration and recrystallized to give 9. This compound was obtained in 63% (3.11 mg) as a brown crystals (from ethanol),m.p. 223-224 ℃. IR (KBr,cm-1) νmax 3392 (NH),2950 (CH-aliph),2206 (C≡N),1702 (C=O). 1H NMR (CDCl3) δ: 1.3 (t,3H,J = 7.1 Hz,CH3),1.82 (m,2H, CH2),2.73 (m,2H,CH2),4.1 (q,2H,J = 7.1 Hz),5.30 (s,1H, pyrimidine-H),6.90-7.56 (m,10H,Ar-H,55CH,NH),11.07 (s,1H, NH,exchangeable with D2O); 13C NMR (75 MHz,DMSO-d6): δ 15.6, 23.5,27.4,58.5,61.6,76.7,116.1,125.1,126.7,128.4,128.8,129.5, 131.8,132.6,133.6,134.1,136.3,138.8,139.6,152.6,163.5,167.6; MS (m/z,%): 492 (M+,26.15),493 (M + 1,18.3),494 (M + 2; 4.9), 447 (4),370 (25),284 (21),244 (22),206 (15),163 (19.5),140 (54), 136 (76),125 (base peak),127 (36),119 (27),76 (7.6). Anal. Calcd. for C26H22Cl2N4O2: C,63.28; H,4.49; N,11.35. Found: C,63.10; H, 4.50; N,11.40.
2.5. 9-(4-Chlorobenzylidene)-6-(4-chlorophenyl)-4-methyl-6,7,8,9- tetrahydrocyclo-penta[d]pyrimido[1,2-a]pyrimidin-2-ol 12To a mixture of compound 3c (0.01 mmol),ethyl acetoacetate (0.01 mmol) in ethanol (30 mL),a few drops of glacial acetic acid was added. The reaction mixture was refluxed for 4 h. The resulting solid product was collected by filtration and recrystallized to give 12. This compound was obtained in 84% (3.66 mg) as a brown crystals (from ethanol),m.p. 200-201 ℃. IR (KBr,cm-1) νmax 3196 (OH),2960 (CH-aliph). 1H NMR (DMSO-d6): δ 1.91 (s,3H,CH3), 2.18 (m,2H,CH2),2.80 (m,2H,CH2),5.21 (s,1H,pyrimidine-H at position-6),7.10 (s,1H,pyrimidine-H at position-3),7.24-7.47 (m, 9H,Ar-H,55CH),8.80 (s,1H,OH,exchangeable with D2O); 13C NMR (75 MHz,DMSO-d6): d 20.4,23.6,26.8,54.3,62.2,124.7,126.7, 128.6,129.4,129.8,132.4,132.9,133.4,134.1,136.0,140.4,142.6, 152.4,156.5,164.4; MS (m/z,%): 435 (M+,9.8),436 (M + 1,4.1),437 (M + 2,5),434 (M-1,5.6),370 (42.5),282 (16.9),246 (30),206 (13.1),140 (base peak),76 (11.6). Anal. Calcd. for C24H19Cl2N3O: C, 66.05; H,4.39; N,9.63. Found: C,66.10; H,4.40; N,9.70.
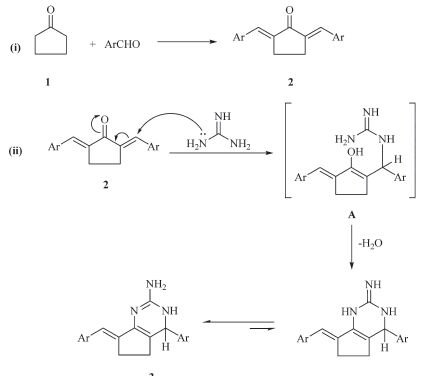
3. Results and discussionTernary condensation of cyclopentanone 1,aromatic aldehyde and guanidine hydrochloride (1:2:1 molar ratio) in ethanol at reflux temperature in the presence of sodium ethoxide furnished the novel cyclopenta[d]pyrimidines 3a-c (Scheme 1). The structures of compounds 3a-c were established on the basis of their elemental analyses and spectral data. The infrared spectra of compounds 3a-c indicated the presence of the NH2 and NH groups. For example,the 1H NMR spectrum of compound 3b (DMSO-d6) showed two multiplet signals at 2.17,2.67 ppm,which were readily assigned to the cyclopentyl protons,a broad singlet at d 3.40 assigned to the amino protons (D2O exchangeable),d 3.72, 3.83,which were assigned for two methoxy protons,d 5.30 assigned to the hydrogen attached at C-4,a singlet signal at d 6.45 assigned to the NH proton (D2O exchangeable) and a multiplet at d 6.82-7.75 assigned to the aromatic and methylideneprotons. The mass spectrum of compound 3c showed a molecular ion peak at m/z = 369 (42.9%) corresponding to the molecular formula C20H17Cl2N3. The base peak was found in the spectrum at m/z = 367 (M-2) and the following fragments were observed in the spectrum at m/z 370 (M + 1; 22%),371 (M + 2; 9.5%),368 (M-1; 98.6%),329 (29%),281 (20%),244 (17%),206 (68%),150 (27%),115 (70%) and 76 (8.7%). In addition,the structure of cyclopenta[d]pyrimidine 3 was established by an independent synthesis by reacting an equimolar amount of 2,5-bis-(arylmethylidene)cyclopentanone 2 [20] with guanidine hydrochloride in methanol in the presence of sodium methoxide. The formation of 3a-c can be explained by the reaction pathway depicted in Scheme 3. The reaction occurs via initial formation of the bis(arylmethylidene)cyclopentanone 2, from the condensation of aromatic aldehyde and cyclopentanone as shown in Scheme 3,which suffer a nucleophilic attack to give the Michael adduct (A). The intermediate (A) then cyclizes, dehydrates and subsequent tautomerizes to give the aminopyrimidine 3 (Scheme 2).

|
Download:
|
| Scheme 1.Multicomponent condensation reaction of cyclopentanone,aromatic aldehyde and guanidine. | |
{kind=link}

|
Download:
|
| Scheme 2.A possible mechanism for the formation of cyclopenta[d]pyrimidines 3. | |
{kind=link}

|
Download:
|
| Scheme 3.Synthetic route to pyrimido[1,2-a]pyrimidine 5 and 7 derivatives. | |
{kind=link}
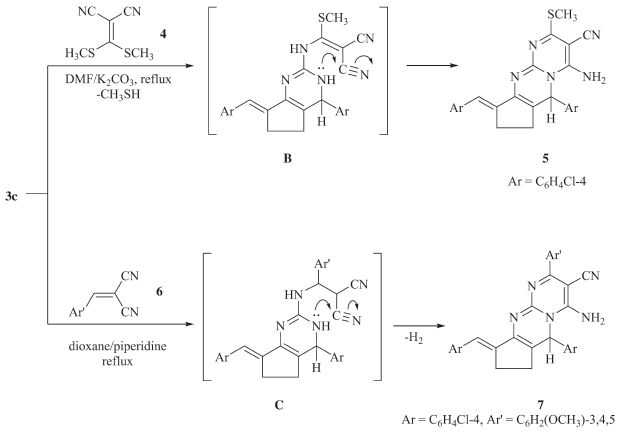
The reactivity of aminopyrimidine derivative 3c toward some electrophilic reagents was studied. It has been found that, treatment of compound 3c with bis(methylthio) methylenemalononitrile 4 in N,N-dimethylformamide under reflux in the presence of anhydrous potassium carbonate gave the novel pyrimido[1,2-a]pyrimidine derivative 5 (Scheme 3). The structure of compound 5 was confirmed on the basis of analytical and spectral data. The infrared spectrum of the reaction product showed the characteristic absorption band at 2207 cm-1 (CN),in addition to amino absorptions at 3319,3196 cm-1. The 1H NMR spectrum (DMSO-d6) of compound 5 revealed a singlet signal at d 2.49 assigned to the methylsulfanyl protons,two signals at d 2.20, 2.90 assigned to cyclopentyl protons,a singlet at d 5.2 assigned to the hydrogen attached at position six of pyrimidopyrimidine and a multiplet at d 6.80-7.6 assigned to aromatic and benzylidene protons in addition to downfield broad singlet at d 8.82 assigned to the amino protons. The formation of 5 is assumed to proceed through intermediate (B),followed by subsequent intramolecular cyclization [21] via a Michael type nucleophilic addition of the amino endocyclic group to the cyano function,Scheme 3. In a similar manner,the reaction of compound 3c with 3,4,5- trimethoxybenzylidenemalononitrile 6 in refluxing dioxane in the presence of catalytic amount of piperidine afforded the novel enaminonitrile derivative 7,via acyclic Michael adduct (C) (Scheme 3). The infrared spectrum of the reaction product displayed absorption bands at 3350,3235 and 2190 cm-1 corresponding to amino and cyano groups,respectively. The mass spectrum of compound 7 showed a molecular ion peak at m/z 612 (M + 1,12.6%),corresponding to the molecular formula C33H27Cl2N5O3.
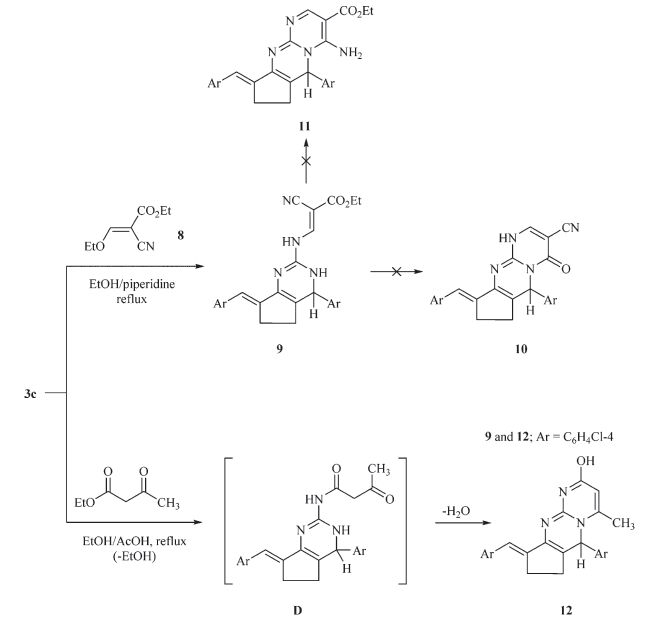
The reaction of compound 3c with ethyl ethoxymethylidenecyanoacetate 8 under reflux in ethanol in the presence of catalytic amounts of piperidine gave aminomethylidenecyanoacetate derivative 9 rather than the expected pyrimidopyrimidines 10 and 11 (Scheme 4). The infrared spectrum of the reaction product revealed absorption bands at 3392,2206 and 1702 cm-1 corresponding to NH,C≡N and C=O groups,respectively. The 1H NMR spectrum (DMSO-d6) showed a triplet at d 1.3 and a quartet at d 4.1 assigned to the ethyl group in addition to the presence of signals for the cyclopentyl,pyrimidine-H,aromatic,benzylidene protons and downfield singlet signal at d 11.07 assigned to the NH proton. Also, the mass spectrum of compound 9 displayed a molecular ion peak at m/z 492 (M+,26.15%) with base peak at m/z 125. The formation of compound 9 is assumed to proceed via condensation of the exocyclic amino group in compound 3c to the activated double bond in ethoxymethylidenecyanoacetate 8,followed by elimination of ethanol molecule,Scheme 4. Refluxing of compound 3c with ethyl acetoacetate in ethanol in the presence of glacial acetic acid furnished the pyrimido[1,2-a]pyrimidine derivative 12 on the basis of analytical and spectral data. The infrared spectrum of compound 12 showed the characteristic absorption band at 3196 cm-1 for the OH group. The 1H NMR spectrum (DMSO-d6) displayed the lack of signals characteristic for ethyl protons and the presence of cyclopentyl,methyl,pyrimidyl,and hydroxyl protons. In addition,the mass spectrum of the reaction product exhibited a molecular ion peak at m/z 435 (9.8%) and the base peak was found in the spectrum at m/z 140. The formation of 12 is assumed to proceed via condensation of the exocyclic amino group [21] of the pyrimidine with ethyl acetoacetate to form (D),followed by intramolecular cyclization by loss of water molecule (Scheme 4).

|
Download:
|
| Scheme 4.Synthetic route to aminopyrimidine 9 and pyrimido[1,2-a]pyrimidine 12 derivatives. | |
{kind=link}
In summary,we synthesized some novel 2-aminopyrimidines from the reaction of cyclopentanone 1,aromatic aldehyde and guanidine (1:2:1 molar ratio) in one-pot reaction in high yields. The reaction of compound 3c with some electrophilic reagents was also investigated.
| [1] | A.R. Katritzky, C.W. Rees, E.F.V. Scriven, Comprehensive Heterocyclic Chemistry II, Pergamon Press, Oxford, 1996. |
| [2] | I.M. Lagoja, Pyrimidines as constituent of natural biologically active compounds, Chem. Biodivers. 2 (2005) 1-50. |
| [3] | (a) H. Kakiya, K. Yagi, H. Shinokubo, et al., Reaction of a, a-dibromo oxime ethers with Grignard reagents: alkylative annulation providing a pyrimidine core, J. Am. Chem. Soc. 124 (2002) 9032-9033; (b) M. Movassaghi, M.D. Hill, Single-step synthesis of pyrimidine derivatives, J. Am. Chem. Soc. 128 (2006) 14254-14255. |
| [4] | G.L. Luo, L. Chen, G.S. Poindexter, Microwave-assisted synthesis of aminopyrimidines, Tetrah. Lett. 43 (2002) 5739-5742. |
| [5] | A.R. Katritzky, B.V. Rogovoys, Recent developments in guanylating agents, Chem. Inform. 36 (2005) 49-87. |
| [6] | W.X. Zhang, Z.M. Hou, Catalytic addition of alkyne C-H, amine N-H, and phosphine P-H bonds to carbodiimides: an efficient route to propiolamidines, guanidines, and phosphaguanidines, Org. Biomol. Chem. 6 (2008) 1720-1730. |
| [7] | C. Alonso-Moreno, A. Antinolo, F. Carrillo-Hermosilla, A. Otero, Guanidines: from classical approaches to efficient catalytic syntheses, Chem. Soc. Rev. 43 (2014) 3406-3425. |
| [8] | W.X. Zhang, L. Xu, Z.F. Xi, Recent development of synthetic preparation methods for guanidines via transition metal catalysis, Chem. Commun. 51 (2015) 254-265. |
| [9] | S. Pirc, D. Bevk, A. Golobič, B. Stanovnik, J. Svete, Transformation of amino acids into nonracemic 1-(heteroaryl)ethanamines by the ignominiousketone methodology, Helv. Chim. Acta 89 (2006) 30-44. |
| [10] | E. Bellur, P. Langer, Synthesis of 4-(3-hydroxyalkyl)pyrimidines by ring transformation reactions of 2-alkylidenetetrahydrofurans with amidines, Tetrahedron 62 (2006) 5426-5434. |
| [11] | D. Hawksley, D.A. Griffin, F.J. Leeper, Synthesis of 3-deazathiamine, J. Chem. Soc. Perkin. Trans. I 1 (2001) 144-148. |
| [12] | N. Singh, S.K. Pandey, N. Anand, et al., Synthesis, molecular modeling and bioevaluation of cycloalkyl fused 2-aminopyrimidines as antitubercular and antidiabetic agents, Bioorg. Medic. Chem. Lett. 21 (2011) 4404-4408. |
| [13] | J. Zhu, H. Bienayme, Multicomponent Reactions, Wiely-VCH, Weinheim, Germany, 2005. |
| [14] | C. Simon, T. Constantieux, J. Rodriguez, Utilisation of 1, 3-dicarbonyl derivatives in multicomponent reactions, Eur. J. Org. Chem. 2004 (2004) 4957-4980. |
| [15] | N. Isambert, M.S. Duque, J.C. Plaquevent, et al., Multicomponent reactions and ionic liquids: a perfect synergy for eco-compatible heterocyclic synthesis, Chem. Soc. Rev. 40 (2011) 1347-1357. |
| [16] | B.B. Touré, D.G. Hall, Natural product synthesis using multicomponent reaction strategies, Chem. Rev. 109 (2009) 4439-4486. |
| [17] | M.S.A. El-Gaby, G.A.M. El-Hag Ali, A.A. El-Maghraby, M.T.A. El-Rahman, M.H.M. Helal, Synthesis, characterization and in vitro antimicrobial activity of novel 2-thioxo-4-thiazolidinones and 4, 4'-bis(2-thioxo-4-thiazolidinone-3-yl)diphenylsulfones, Eur. J. Med. Chem. 44 (2009) 4148-4152. |
| [18] | M.S.A. El-Gaby, Z.H. Ismail, S.M. Abdel-Gawad, H.M. Aly, M.M. Ghorab, Synthesis of thiazolidinone and thiophene derivatives for evaluation as anticancer, Phosph., Sulf. Silic. Rel. Elem. 184 (2009) 2645-2654. |
| [19] | M.S.A. El-Gaby, S.I. Mohamed, H.A. Eyada, et al., New approach for the synthesis of pyrano[2, 3-d]thiazoles, J. Mater. Sci. Eng. A1 (2011) 705-710. |
| [20] | M.S. Abaee, M.M. Mojtahedi, S. Forghani, et al., A green, inexpensive and efficient organocatalyzed procedure for aqueous aldol condensations, J. Braz. Chem. Soc. 20 (2009) 1895-1900. |
| [21] | M.S.A. El-Gaby, A.M. Hussein, A.A. El-Adasy, et al., Synthesis and biological evaluation of novel substituted furan, pyrimidine and pyrimido[1,2-a]pyrimidine derivatives having diphenyl sulfide moiety, Int. J. Pharm. Sci. 4 (2014) 780-786. |