




b Department of Pharmacy, Wenzhou Medical University, Wenzhou 325000, China;
c Jiangsu Key Laboratory for Biomaterials and Devices, School of Biological Science and Medical Engineering, Southeast University, Nanjing 210008, China
Epigenetics is widely implicated in tumor initiation and progression by different modifications of DNA and histones [1, 2]. There is a growing body of evidence demonstrating the importance of histone modification in silencing tumor suppressor genes [3]. Histone acetylation and deacetylation play crucial roles for regulating protein function of eukaryotic cells,which are correlated with two classes of enzymes: [7TD$DIF]Histone acetyltransferases (HATs) and histone deacetylases (HDACs) [4]. HATs add acetyl groups to lysine residues of histone tails causing localized relaxation of chromatin and transcriptional activation of nearby genes,while HDACs remove the acetyl groups of acetylated histones leading to transcriptional repression [4, 5]. The maintenance of equilibrium between acetylation and deacetylation of histones and non-histone substrates is essential for normal cell growth. However,HDAC overexpression has been found in a variety of human cancers and inflammation,including myeloid neoplasia and solid tumors [6]. HDACs play important roles in the upstream control of gene transcription,cell cycle progression,andapoptosis. Consequently,it has been widely recognized that HDACs are promising targets for intervention of a number of cancers [7, 8, 9].
It is known that histone deacetylase inhibitors (HDACi) have been shown to induce cell growth arrest,differentiation and/or apoptosis in different cancer cell lines. More encouragingly,the suberoylanilide hydroxamic acid (SAHA,Vorinostat,Fig. 1) has been licensed for the treatment of cutaneous T cell lymphoma treatment (CTCL) on 2006 [10]. In November 2009,FDA also approved romidepsin (FK228) for treatment of CTCL in patients who have received at least one prior systemic therapy [11]. These two approved drugs have validated the therapeutic use of HDAC inhibitors in cancer therapy.

|
Download:
|
| Fig. 1.Structures of representative HDAC inhibitors and target compounds 8a-8o. | |
There are a number of new HDACi that are currently undergoing various stages of clinical development for therapy of multiple cancer types [9, 12, 13]. The common pharmacophore of these HDACi consists of three domains: A zinc-binding group (ZBG),such as hydroxamic acid; a cap group,generally a hydrophobic and aromatic group; a saturated or unsaturated linker domain, composed of linear or cyclic structures that connect the ZBG and the cap group [14]. HDAC inhibitors are grouped chemically into four classes: Hydroxamic acids,benzamides,short-chain fatty acids,and cyclic tetrapeptides [15]. To date,many studies on HDACi development have focused on the arylhydroxamic acids. Among these N-hydroxyarylamide derivatives,Belinostat(PXD101),pracinostat (SB939),and dacinostat (LAQ824) in clinical trials share a common active fragment N-hydroxycinnamamide (see Fig. 1),and exhibited excellent HDAC inhibitory activity [16- 18]. In addition,abexinostat (PCI-24781) and givinostat (ITF2357) with an N-hydroxybenzamide moiety are currently undergoing clinical trials [19, 20].
Although HDACis showed potent antitumor effects,they also exhibited side effects that might limit their clinical potential [21]. Therefore,looking for new HDACis that are specific-inhibition to one kind HDAC subtype is extremely urgent and necessary. As part of our ongoing effort to discover novel anticancer agents,we were inspired by the fact that many natural or synthetic products bearing indole rings usually display excellent antiproliferative activities against several cancer cells in recent years [22, 23]. More importantly,some HDACis containing the indole ring as cap group also show prominently the clinical therapeutic effect of cancer [18, 24]. In addition,it is well-known that click chemistry has been widely applied in organic synthesis and drug discovery to afford a highly efficient combinatorial approach to construct a library of new HDACi candidates [25, 26]. And the triazole ring is often used as a cap group,which may have favorable π-π stacking interactions with phenylalanine residues of HDACs. With these ideas in mind,the novel target compounds 8a-8o were designed and synthesized by connecting indole ring with N-hydroxyarylamide through alkyl substituted triazole. We proposed that the triazole ring could act as a linking moiety which joins the cap group to the linker group in the structure of HDACi. Here we report the synthesis and biological evaluation of the target compounds.
2. ExperimentalMelting points were determined on a RDCSY-I capillary apparatus and were uncorrected. The compounds synthesized were purified by column chromatography using silica gel (200- 300 mesh) except for recrystallization and thin-layer chromatography (TLC) using silica gel 60 F254 plates (250 mm; Qingdao Ocean Chemical Company,China). 1H NMR and 13C NMR spectra were recorded with a Bruker Avance spectrometer at 300 K,using TMS as an internal standard. MS spectra were recorded on a Mariner Mass Spectrum (ESI) and HRMS on Agilent technologies LC/MSD TOF.
The synthetic route of these target compounds 8a-8o is outlined in Scheme 1. The key reaction in the synthesis of the target compounds is the click chemistry between azides and terminal alkynes [27]. On the one hand,the indole bromide 1a-1c were reacted with NaN3 to generate indole azides 2a-2c in MeCN solution. On the other hand,various hydroxyl substituted methyl cinnamate 3a-3e were treated with 3-bromoprop-1-yne to obtain etherified alkynes 4a-4e,which subsequently were reacted with indole azides 2a-2c to give key intermediates 5a-5o in the presence of CuSO4·5H2O and sodium ascorbate by click chemistry. Next,intermediates 5a-5o were subsequently hydrolyzed to obtain 6a-6o,which were directly treated with tetrahydro-2Hpyran- 2-ol to form compounds 7a-7o in the presence of Nmethylmorpholine and ethyl chloroformate. Finally,O-protected groups of 7a-7o were removed by CF3COOH (TFA) to provide target compounds 8a-8o. The products 8a-8o were purified by column chromatography,and their structure data of MS,1H NMR spectra and HRMS of selected compounds were shown in reference [28].

|
Download:
|
| Scheme 1.Reagents and conditions: (a) NaN3,MeCN,reflux,3 h,81%-85%. (b)K2CO3,KI,CH3CN,reflux,3-6 h,72%-78%.(c) CuSO4·5H2O,sodium ascorbate,DMF,80 ℃,3-5 h, 66%-75%. (d) 1 mol/L NaOH (aq.),MeOH,r.t.,2-4 h,89%-93%.(e)1.Ethyl chloroformate,N-methylmorpholine,THF,0℃ to r.t.,1 h; 2.tetrahydro-2H-pyran-2-ol,TEA,THF,0℃,1 h,53%-66%. (f) TFA,CH2Cl2,r.t.,2-4 h,82%-89%. | |
General synthetic procedure for 2a-2c: A mixture of indole bromide 1 (10 mmol) and NaN3 (0.78 g,12 mmol) in 50 mL acetonitrile was refluxed for 3 h. The solvent was evaporated in vacuum and water was added. The solution was then extracted with ethyl acetate (50 mL,3×). The organic layer was concentrated in vacuo to yield 2a-2c as yellowish oil (81%-85%).
General synthetic procedure for 4a-4e: A mixture of 3a-3c (5 mmol),K2CO3 (0.7 g,5 mmol) and KI (83 mg,0.5 mmol) in acetonitrile (40 mL) was refluxed and stirred for 3-6 h. The solvent was evaporated in vacuum and water was added. The solution was then extracted with ethyl acetate (40 mL,3×). The organic layer was combined,washed with brine,dried with anhydrous Na2SO4 and concentrated in vacuo,affording 4a-4e as pale yellow solid (72%-78%).
General synthetic procedure for 5a-5o: A mixture of 3a-3c (3 mmol),CuSO4·5H2O (0.30 g,1.2 mmol) and sodium ascorbate (0.36 g,1.8 mmol) in DMF (40 mL) was stirred for 3-5 h at 80 ℃. The solution was then cooled,and poured into 200 mL water, which was subsequently extracted with ethyl acetate (50 mL,3×). The organic layer was combined,washed with brine,dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (petroleum ether-ethyl acetate = 5:1-2:1,v/v as the eluent) to give 5a-5o as white solid (66%-75%).
General synthetic procedure for 7a-7o: To a solution of 5a-5o (1.2 mmol) in methanol (20 mL),1 mol/L NaOH (3.2 mL) was dropwised added. The mixture was refluxed for 2-4 h,and then cooled. The solvent was evaporated and the residue was dissolved in water. The mixture was acidified with 2 mol/L HCl solution to pH 5. The precipitate was filtered,washed with water,and dried in vacuum to afford the crude,which was subsequently added into a solution of N-methylmorpholine (0.12 g,1.4 mmol) and ethyl carbonochloridate (0.13 g,1.2 mmol) in 20 mL anhydrous THF at 0 ℃. Next,the mixture was stirred at room temperature for 1 h, which was then added dropwise to a solution of tetrahydro-2Hpyran- 2-ol (0.14 g,8 mmol) in 5 mL anhydrous THF. After the reaction was completed,the resulting mixture was allowed to pour into ice-water,and extracted with ethyl acetate (20 mL,3×). The organic phase was washed with water and brine,then dried over anhydrous sodiumsulfate,filtered and evaporated to afford the crude product,which was purified by column chromatography on silica gel to give compound 7a-7o as pale yellow waxy solid (53%-66%).
General synthetic procedure for 8a-8o: A solution of 7a-7o (0.70 mmol) and CF3COOH (1 mL) in 5 mL dry CH2Cl2 was stirred at room temperature for 2 h. The solvent was removed under reduced pressure. The crude residue was dissolved in 10 mL dichloromethane and 2 mL Et3N was slowly added to the solvent,and thecrude product was purified by column chromatography (MeOH/CH2Cl2 = 1:5-1:8) to yield 8a-8o as white waxy solid (82%-89%).
3. Results and discussionThe anti-proliferation inhibitory activities of target compounds 8a-8o against five human cancer cells HCT-116,Lovo (human colon carcinoma cells),K562 (human leukemia cells),MCF-7(human breast adenocarcinoma cells),and Hep G2 (human hepatocellular carcinoma cells) were evaluated by MTT assays in vitro,with SAHA as positive control. The values of half inhibitory concentration (IC50) about all the target compounds against each tumor cell line were measured and presented in Table 1. Most of the target compounds displayed good to moderate antiproliferative activities. Moreover,among these compounds,compound 8n exhibited strongest antiproliferative activities with IC50 values of 3.57-6.21 μmol/L against each tested cancer cell,which were comparable to or slightly stronger than SAHA (IC50 = 4.95- 7.11 μmol/L). Interestingly,compound 5n (the intermediate of 8n ) showed weak cancer inhibitory activities,which were 3-4-fold less than those of 8n . Such phenomenon suggested that the hydroxamic acid moiety has a positive influence on the cytotoxicity of this series of N-hydroxyarylamide derivatives.
|
|
Table 1 The IC50 values of 5n and 8a-8o against five human cancer cell lines |
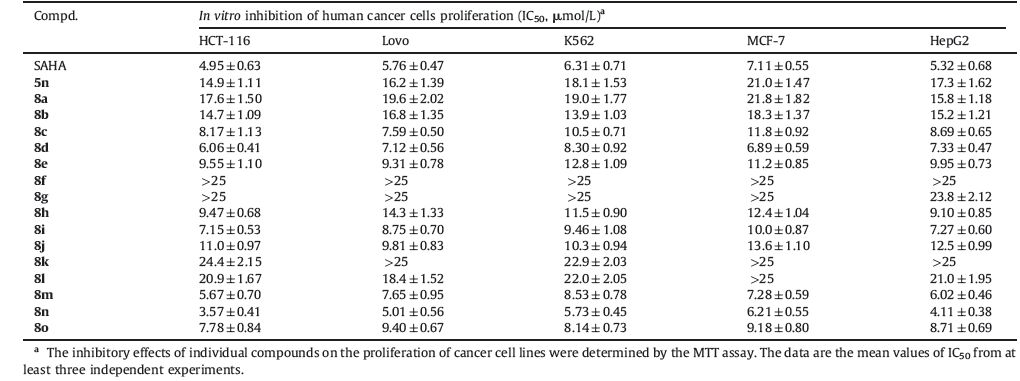
Furthermore,in order to explore the HDAC inhibitory activity and isoform selectivity profile,the active compounds 8c-8d,8i, and 8m-8o were chosen to conduct enzyme inhibitory effect against HDAC1,HDAC6,and HDAC8 based on fluorescent-based HDAC biochemical activity assay. Results listed in Table 2 showed that 8c-8d,8i,and 8m-8o also exhibited promising HDAC1, HDAC6,and HDAC8 inhibitory activities,especially for HDAC1, which were comparable to or slightly better than SAHA. Apparently,the HDACs inhibitory effects of individual compounds were associated with their cytotoxicities against these cancer cells in vitro. What is more,compounds 8c,8d,8m,and 8n exhibited obvious selectivity against HDAC1,among which compound 8n exhibited the most prominent selectivity. IC50 value of 8n against HDAC1 was 0.078 mmol/L,which was nearly four-fold and fivefold lower than that of HDAC6 and HDAC8,respectively. Thepositive control,SAHA,had almost no selectivity toward these HDAC isoforms.
|
|
Table 2 The HDACs inhibitory activities of 8c-8d,8i,and 8m-8o |

{kind=link}
{kind=link}
Analysis of SAR revealed that 8a-8o with different Nhydroxyarylamide, indole ring and the lengths of the alkyl triazoles displayed variable antitumor activities. Particularly for N-hydroxyarylamide moiety which was crucial for their anticancer activity in vitro,N-hydroxycinnamamide derivatives 8m-8o demonstrated more potent antiproliferative activities than Nhydroxybenzamide derivatives 8k-8l against human cancer cells. Furthermore,the compounds 8k-8o linked with 5-methoxy indole ring exhibited slightly stronger cancer cell inhibitory effects than the other derivatives 8a-8j. In addition,variations in the carbochain lengths of substituted triazoles also affected the in vitro anticancer activity of these derivatives. For example,the compounds 8a-8e and 8k-8o with a one-carbon linked triazole showed relatively stronger anticancer activity than 8f-8j with a two-carbon linker. The plausible reason may be that the onecarbon linked triazole ring could be of suitable size to fit into the narrow active pocket ofHDAC protein. This indicated that,besides the role of N-hydroxyarylamide and indole ring fragments,the triazole ring in our design also contributed to the antitumor activities.
Since tumor cells at cell cycling arrest are associated with HDAC inhibitory effect,thus,the impact of 8n on colon cancer cell cycling was examined. The HCT-116 cells were incubated with vehicle alone or with different concentrations of 8n (1.8 or 3.6 mmol/L) for 48 h,and the percentages of the G0/G1,S,or G2/M cells were determined by PI staining and flow cytometry (see Fig. 2). It was found that compared with control,treatment with 8n dosedependently induced the colon cancer cell cycling arrest at G0/G1 phase in vitro.

|
Download:
|
| Fig. 2.Effects of 8n on cell-cycling of human colon cancer HCT-116 cells. (A) Cells were treated with 8n and stained with PI,followed by flow cytometry analysis. (B) Data are representative histograms and expressed as the means±SD. | |
{kind=link}
To get insight into the preliminarily molecular mechanism underlying the HDAC inhibition in 8n -treated cells,we conducted western blot analysis to examine the expression of the acetylation for histone H3 and tubulin in HCT-116 cells. The HCT-116 cells were incubated with the vehicle alone,8n (1.8 or 3.6 μmol/L),or SAHA (5.0 μmol/L). The acetylation levels of histone H3 and tubulin were determined by immunoblotting assays using b-actin as the control (Fig. 3). It was observed that 8n dramatically enhanced the acetylation of histone H3 and tubulin in a dosedependent manner,which was comparable to or slightly stronger than that of SAHA (5 μmol/L). The results demonstrated that 8n can effectively inhibit HDAC by promoting the expression of the acetylation for histone H3 and tubulin in vitro.

|
Download:
|
| Fig. 3.Immunoblot analysis of the expression of the acetylation for histone H3 and tubulin in vitro. The expression of Ac-H3,Ac-tubulin,and b-actin were examined by western blot analysis. HCT-116 cells were incubated with or without 8n,or SAHA at the indicated concentrations for 48 h and the levels of protein expression were detected using specific antibodies. Data shown are representative images of each protein for three separate experiments. | |
{kind=link}
Insummary,a series of novel hybrids 8a-8o were discovered and synthesized by connecting indole ring with N-hydroxyarylamide through click chemistry,and their biological activities were evaluated. The in vitro assay of their inhibitory activities against five cancel cell lines showed thatmost target compounds displayed good to moderate ability to inhibit multiple cancer cells growth. Especially,compound 8n had significant antiproliferation and HDACs inhibitory activities,comparable to or slightly stronger than SAHA,in human carcinoma cells. What is important,compound 8n showed features of high potency and selectivity of HDAC1,which was nearly four-fold and five-fold lower than that of HDAC6 and HDAC8,respectively. In addition,compound 8n also could dosedependently induce cancer cell cycling arrest at G0/G1 phase and promote the expression of the acetylation for histoneH3 and tubulin in vitro. Thiswork could serve as a foundation for further exploration of selective HDAC inhibitors for the treatment of cancer.
| [1] | Z. Li, W.G. Zhu, Targeting histone deacetylases for cancer therapy: from molecular mechanisms to clinical implications, Int. J. Biol. Sci. 10 (2014) 757-770. |
| [2] | C.B. Yoo, P.A. Jones, Epigenetic therapy of cancer: past, present and future, Nat. Rev. Drug Discov. 5 (2006) 37-50. |
| [3] | L. Simó-Riudalbas, M. Esteller, Targeting the histone orthography of cancer: drugs for writers, erasers and readers, Br, J. Pharmacol. 172 (2015) 2716-2732. |
| [4] | H. Lehrmann, L.L. Pritchard, A. Harel-Bellan, et al., Histone acetyltransferases and deacetylases in the control of cell proliferation and differentiation, Adv. Cancer Res. 86 (2002) 41-65. |
| [5] | B.E. Bernstein, J.K. Tong, S.L. Schreiber, Genome wide studies of histone deacetylase function in yeast, Proc. Natl. Acad. Sci. U. S. A. 97 (2000) 13708-13713. |
| [6] | O. Witt, H.E. Deubzer, T. Milde, I. Oehme, et al., HDAC family: what are the cancer relevant targets, Cancer Lett. 277 (2009) 8-21. |
| [7] | P. Bose, Y. Dai, S. Grant, Histone deacetylase inhibitor (HDACI) mechanisms of action: emerging insights, Pharmacol. Ther. 143 (2014) 323-336. |
| [8] | A.C. West, R.W. Johnstone, New and emerging HDAC inhibitors for cancer treatment, J. Clin. Invest. 124 (2014) 30-39. |
| [9] | M. Slingerland, H.J. Guchelaar, H. Gelderblom, Histone deacetylase inhibitors: an overview of the clinical studies in solid tumors, Anticancer Drugs 25 (2014) 140-149. |
| [10] | P.A. Marks, Discovery and development of SAHA as an anticancer agent, Oncogene 26 (2007) 1351-1356. |
| [11] | FK228: http://www.fda.gov/NewsEvents/Newsroom/Press Announcements/2009/ucm189629.htm. |
| [12] | T. Qiu, L. Zhou, W. Zhu, et al., Effects of treatment with histone deacetylase inhibitors in solid tumors: a review based on 30 clinical trials, Future Oncol. 9 (2013) 255-269. |
| [13] | T. You, K. Chen, F.H. Wang, et al., Design, synthesis, and biological evaluation of Nhydroxycinnamamide/salicylic acid hybrids as histone deacetylase inhibitors, Chin. Chem. Lett. 25 (2014) 474-478. |
| [14] | T.A. Miller, D.J. Witter, S. Belvedere, Histone deacetylase inhibitors, J. Med. Chem. 46 (2003) 5097-5116. |
| [15] | P.A. Marks, The clinical development of histone deacetylase inhibitors as targeted anticancer drugs, Expert Opin. Invest. Drugs 19 (2010) 1049-1066. |
| [16] | J. McDermott, A. Jimeno, Belinostat for the treatment of peripheral T-cell lymphomas, Drugs Today (Barc.) 50 (2014) 337-345. |
| [17] | W.P. Yong, B.C. Goh, R.A. Soo, et al., Phase I and pharmacodynamic study of an orally administered novel inhibitor of histone deacetylases, SB939, in patients with refractory solid malignancies, Ann. Oncol. 22 (2011) 2516-2522. |
| [18] | J.S. de Bono, R. Kristeleit, A. Tolcher, et al., Phase I pharmacokinetic and pharmacodynamic study of LAQ824, a hydroxamate histone deacetylase inhibitor with a heat shock protein-90 inhibitory profile, in patients with advanced solid tumors, Clin. Cancer Res. 14 (2008) 6663-6673. |
| [19] | S. Fouliard, R. Robert, A. Jacquet-Bescond, et al., Pharmacokinetic/pharmacodynamic modelling-based optimisation of administration schedule for the histone deacetylase inhibitor abexinostat (S78454/PCI-24781) in phase I, Eur. J. Cancer 49 (2013) 2791-2797. |
| [20] | A. Furlan, V. Monzani, L.L. Reznikov, et al., Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (givinostat), Mol. Med. 17 (2011) 353-362. |
| [21] | Ø. Bruserud, C. Stapnes, E. Ersvaer, et al., Histone deacetylase inhibitors in cancer treatment: a review of the clinical toxicity and the modulation of gene expression in cancer cell, Curr. Pharm. Biotechnol. 8 (2007) 388-400. |
| [22] | J. Amato, N. Iaccarino, B. Pagano, et al., Bis-indole derivatives with antitumor activity turn out to be specific ligands of human telomeric G-quadruplex, Front. Chem. 2 (2014) 54. |
| [23] | M.T. Macdonough, T.E. Strecker, E. Hamel, et al., Synthesis and biological evaluation of indole-based, anti-cancer agents inspired by the vascular disrupting agent 2-(30-hydroxy-40-methoxyphenyl)-3-(300,400,500-trimethoxybenzoyl)-6-methoxyindole (OXi8006), Bioorg. Med. Chem. 21 (2013) 6831-6843. |
| [24] | Y.S. Cho, L. Whitehead, J. Li, et al., Conformational refinement of hydroxamatebased histone deacetylase inhibitors and exploration of 3-piperidin-3-ylindole analogues of dacinostat (LAQ824), J. Med. Chem. 53 (2010) 2952-2963. |
| [25] | C.D. Hein, X.M. Liu, D. Wang, Click chemistry, a powerful tool for pharmaceutical sciences, Pharm. Res. 25 (2008) 2216-2230. |
| [26] | G.C. Tron, T. Pirali, R.A. Billington, et al., Click chemistry reactions in medicinal chemistry: applications of the 1,3-dipolar cycloaddition between azides and alkynes, Med. Res. Rev. 28 (2008) 278-308. |
| [27] | A.R. Bogdan, K. James, Efficient access to new chemical space through flowconstruction of druglike macrocycles through copper-surface-catalyzed azidealkyne cycloaddition reactions, Chemistry 16 (2010) 14506-14512. |
| [28] | The data of selected compounds: 8c: Yield: 86%, mp 116-119℃; ESI-MSm/z: 390[M+H]+. 1H NMR (300 MHz, DMSO-d6):δ5.31 (s, 2H), 5.43 (s, 2H), 6.55 (d, 1H, J = 15.9 Hz), 6.96-6.99 (m, 2H), 7.08-7.11 (m, 2H), 7.15 (d, 1H, J = 2.4 Hz), 7.35 (m, 1H), 7.57 (d, 1H, J = 7.8 Hz), 7.63-7.68 (m, 3H), 7.79 (s, 1H), 8.96 (s, 1H), 10.41 (s, 1H). 13C NMR (75 MHz, DMSO-d6):δ52.5, 71.2, 107.4, 111.8, 115.6, 118.2, 118.5, 119.3, 120.7, 122.7, 123.1, 127.1, 130.5, 136.0, 139.2, 146.3, 155.1, 164.6. ESIHRMS (m/z): [M+H]+ calcd. for C21H20N5O3: 390.1566; found: 390.1558. 8d: Yield: 83%, mp 123-125℃; ESI-MS m/z: 420 [M+H]+. 1H NMR (300 MHz, DMSO-d6):δ3.81 (s, 3H), 5.29 (s, 2H), 5.50 (s, 2H), 6.53 (d, 1H, J = 15.9 Hz), 6.94-6.97 (m, 3H), 7.08-7.10 (m, 2H), 7.15-7.21 (m, 3H), 7.33 (d, 1H, J = 7.8 Hz), 7.56 (d, 1H, J = 7.8 Hz), 7.62 (d, 1H, J = 15.9 Hz), 7.77 (s, 1H), 8.91 (s, 1H), 10.35 (s, 1H). 13C NMR (75 MHz, DMSO-d6):δ52.0, 56.3, 71.5, 107.7, 110.7, 111.6, 114.4, 118.3, 118.9, 119.5, 121.0, 122.9, 123.3, 127.6, 136.3, 138.9, 146.2, 146.5, 154.8, 164.7. ESI-HRMS (m/z): [M+H]+ calcd. for C22H22N5O4: 420.1672; found: 420.1665. 8i: Yield: 89%, mp 105-108℃; ESI-MS m/z: 434 [M+H]+. 1H NMR (300 MHz, DMSO-d6):δ2.61 (t, 2H, J = 6.6 Hz), 3.37 (t, 2H, J = 6.6 Hz), 3.83 (s, 3H), 5.33 (s, 2H), 6.56 (d, 1H, J = 15.9 Hz), 6.92-6.98 (m, 3H), 7.09-7.12 (m, 2H), 7.17-7.22 (m, 3H), 7.35 (d, 1H, J = 7.8 Hz), 7.57 (d, 1H, J = 7.8 Hz), 7.65 (d, 1H, J = 15.9 Hz), 7.80 (s, 1H), 8.95 (s, 1H), 10.39 (s, 1H). 13C NMR (75 MHz, DMSO-d6):δ28.1, 50.3, 56.5, 71.3, 106.9, 110.5, 111.3, 114.5, 117.8, 118.5, 119.2, 120.7, 122.5, 123.1, 127.3, 136.3, 138.9, 146.2, 146.7, 155.1, 164.9. ESI-HRMS (m/z):[M+H]+ calcd. for C23H24N5O4: 434.1828; found: 434.1842. 8m: Yield: 82%, mp 121-124℃; ESI-MS m/z: 420 [M+H]+. 1H NMR (300 MHz, DMSO-d6):δ3.80 (s, 3H), 5.30 (s, 2H), 5.46 (s, 2H), 6.52 (d, 1H, J = 15.9 Hz), 6.76 (m, 1H), 6.96 (d, 2H, J = 8.4 Hz), 7.11-7.15 (m, 2H), 7.22 (d, 1H, J = 7.8 Hz), 7.61-7.66 (m, 3H), 7.75 (s, 1H), 8.87 (s, 1H), 10.25 (s, 1H). 13C NMR (75 MHz, DMSO-d6):δ52.1, 56.8, 71.5, 102.1, 111.9, 112.2, 115.5, 118.4, 122.6, 123.3, 128.3, 129.1, 130.1, 138.9, 146.2, 146.6, 155.3, 165.1. ESI-HRMS (m/z): [M+H]+ calcd. for C22H22N5O4: 420.1672; found: 420.1681. 8n: Yield: 85%, mp 110-112℃; ESI-MS m/z: 450 [M+H]+. 1H NMR (300 MHz, DMSO-d6):δ3.83 (s, 6H), 5.35 (s, 2H), 5.53 (s, 2H), 6.57 (d, 1H, J = 15.9 Hz), 6.79 (m, 1H), 6.95-7.00 (m, 3H), 7.12-7.23 (m, 5H), 7.65 (d, 1H, J = 15.9 Hz), 7.81 (s, 1H), 8.97 (s, 1H), 10.41 (s, 1H). 13C NMR (75 MHz, DMSO-d6):δ52.3, 56.8, 71.6, 101.9, 110.3, 111.1, 112.1, 112.7, 118.5, 122.5, 123.2, 128.9, 129.3, 130.4, 138.7, 146.2, 146.4, 146.8, 154.7, 164.7. ESI-HRMS (m/z): [M+H]+ calcd. for C23H24N5O5: 450.1777; found: 450.1786. 8o: Yield: 83%, mp 118-120℃; ESI-MS m/z: 420 [M+H]+. 1H NMR (300 MHz, DMSO-d6):δ3.79 (s, 3H), 5.32 (s, 2H), 5.45 (s, 2H), 6.50 (d, 1H, J = 15.9 Hz), 6.88-6.93 (m, 2H), 7.09-7.17 (m, 4H), 7.24 (d, 1H, J = 7.8 Hz), 7.53 (m, 1H), 7.62 (d, 1H, J = 15.9 Hz), 7.73 (s, 1H), 8.82 (s, 1H), 10.33 (s, 1H). 13C NMR (75 MHz, DMSO-d6):δ52.8, 71.5, 102.0, 112.0, 112.4, 114.7, 118.5, 122.7, 123.1, 128.0, 128.5, 130.2, 137.6, 146.2, 146.4, 155.3, 164.9. ESI-HRMS (m/z): [M+H]+ calcd. for C22H22N5O4: 420.1672; found: 420.1659. |