




Primary (hetero)aryl amines are widely used in the synthesis of natural products,pharmaceuticals,agrochemicals as well as polymers and materials [1]. The common methods for preparation of primary amines include coupling of aryl halides with ammonia [2],reductive amination of carbonyl compounds [3],and hydroamination of alkenes [4, 5, 6]. Recently,ammonia,as one of the most attractive sources of nitrogen,has attracted a lot of attentions due to its great abundance and extremely low cost [7, 8]. Very recently, a few methodological advancements for coupling aryl halides with aqueous ammonia to deliver aryl primary amines under mild conditions have been developed [9, 10].
Aryl sulfonates that are easily prepared,usually crystalline,and lower toxicity,are with potential values to investigate as better materials to synthesize primary amines. Despite great progress toward the preparation of primary amines has been made,selective synthesis of primary amines from ammonia still encounters challenges,i.e. requirement of transition-metal,overreactions of primary amines with ammonia. Hence,further efforts were needed to developing a metal-free,mild method for the selective synthesis of primary amines directly from aqueous ammonia.
3,4-Dihydropyrimidinones and their derivatives have consequently been extensively used as a drug-like scaffold [11] and utilization as important precursors in the synthesis of pyrimidine bases [12]. In continuation of our ongoing interest in the synthesis of 3,4-dihydropyrimidinone derivatives [13],we are recently interesting in the synthesis of 2-aminopyrimidines.
2-Aminopyrimidines show interesting biological activities such as inhibitors of rhoassociated protein kinease [14, 15],glycogen synthase kinease 3 (GSK3) [16],and of N-type calcium channels [17]. Notably,the 2-amino-4-arylpyrimidine heterocycle is also found in important drugs such as the hypocholesterolemic agent rosuvastatin [18, 19] and the potent anticancer drug Gleevec [20].
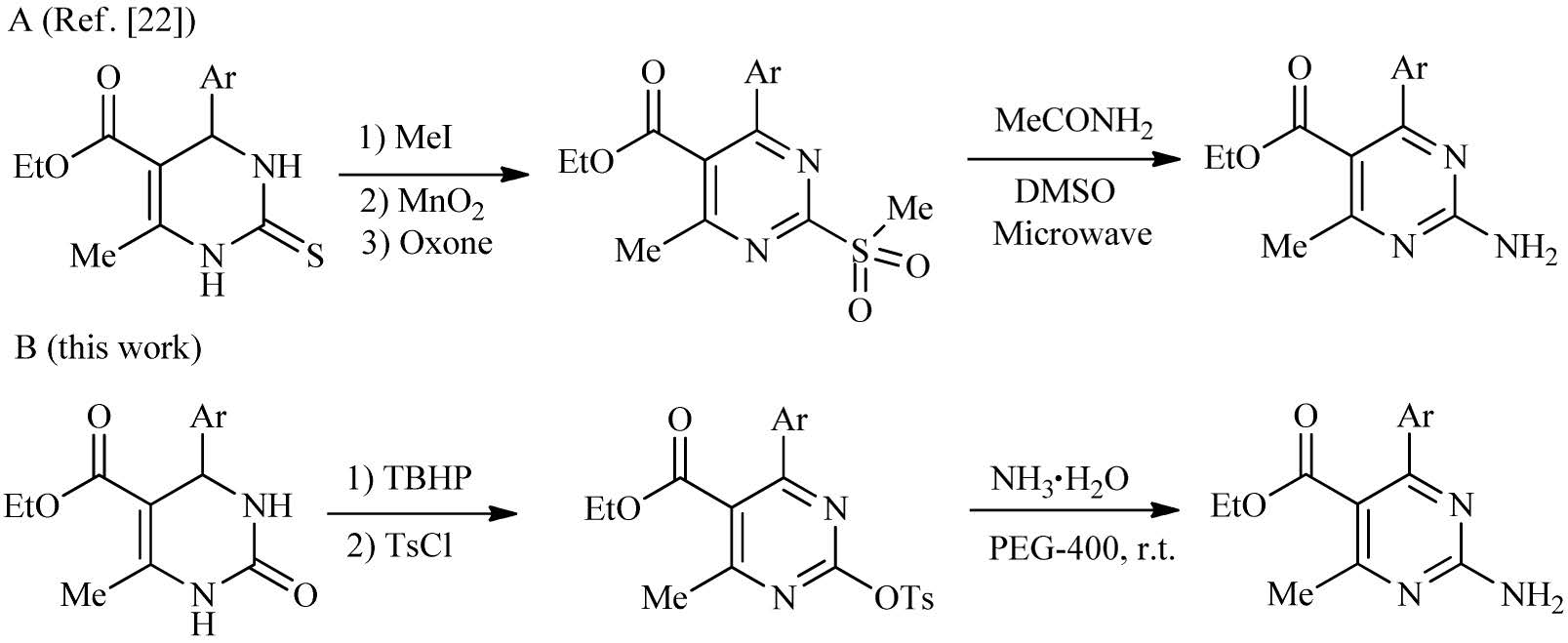
Usually,2-aminopyrimidine subunits are constructed by condensation reactions of enones with corresponding guanidine or nitrogen-containing building blocks [21]. In 2007,Kappe et al. [22] have described a three-step procedure to convert Biginelli DHPMs to 2-methylsulfonyl-pyrimidines,which subsequently converted to 2-aminopyrimidine by the substitution of the reactive sulfonyl group with ammonium acetate as substitute for NH3 (Scheme 1,Method A).

|
Download:
|
| Scheme 1.Synthesis of the 2-aminopyrimidines starting from 3,4-dihydropyrimidinones. | |
Herein we developed a metal-free approach for the synthesis of 2-aminopyrimidines directly from pyrimidin-2-yl tosylates with aqueous ammonia under mild conditions in PEG medium (Scheme 1,Method B).
2. ExperimentalCommercially available reagents were used without further purification unless otherwise stated. Melting points were measured on a XT-4 apparatus and are uncorrected. NMR spectra were recorded at 400 MHz (1H) and 100 MHz (13C),respectively,on a Varian Mercury plus-400 instrument using CDCl3 as solvent and TMS as internal standard. High-resolution mass spectra (HRMS) were obtained on a Bruker Daltonics APEX II 47e mass spectrometer. Column chromatography was generally performed on silica gel (200-300 mesh) and TLC inspections were on silica gel GF254 plates.
2.1. General procedure for the synthesis of 2-amino pyrimidines (2a- 2n)The pyrimidin-2-yl tosylate (1,1.0 mmol),PEG-400 (2 mL) and ammonia water (10 mmol) were added into a test tube. The tube was then sealed with a balloon,and the mixture was stirred at r.t. for 24 h. Then the mixture was poured into water to precipitate the product. Crude product was obtained by means of vacuum filtration,and was further purified by column chromatography on silica gel with petroleum ether/ethyl acetate (3:1) and (1:1) to give the corresponding products 2a-i and 2j-n, respectively.
Ethyl 2-amino-4-methyl-6-phenylpyrimidine-5-carboxylate (2a): White solid,mp 132-133 ℃ [22]. 1H NMR (400 MHz,CDCl3): δ 7.52-7.50 (m,2H),7.41 (d,3H,J = 5.2 Hz),5.82 (s,2H),4.05 (q,2H, J = 7.2 Hz),2.48 (s,3H),0.94 (t,3H,J = 7.6 Hz); 13C NMR (100 MHz, CDCl3): δ 168.31,167.48,166.47,161.98,138.60,129.35,128.15, 127.63,115.95,61.00,22.58,13.40.
Ethyl 2-amino-4-(4-fluorophenyl)-6-methylpyrimidine-5-carboxylate (2b): White solid,mp 167-168 ℃. 1H NMR (400 MHz, CDCl3): δ 7.53-7.49 (m,2H),7.08 (t,2H,J = 8.6 Hz),5.82 (d,2H, J = 10.0 Hz),4.07 (q,2H,J = 7.2 Hz),2.48-2.40 (m,3H),1.00 (t,3H, J = 7.1 Hz); 13C NMR (100 MHz,CDCl3): δ 168.30,167.60,164.98 (d, J = 38.0 Hz),162.31,161.92,134.70,129.84 (d,J = 8.0 Hz),116.14, 115.27 (d,J = 22.0 Hz),61.16,22.65,13.58; HRMS: calcd. for C14H15FN3O2 [M+H]+: 276.1143; found 276.1147.
Ethyl 2-amino-4-(4-chlorophenyl)-6-methylpyrimidine-5-carboxylate (2c): White solid,mp 164-166 ℃. 1H NMR (400 MHz, CDCl3): δ 7.46 (d,2H,J = 8.4 Hz),7.38 (d,2H,J = 8.4 Hz),5.74 (s,2H), 4.08 (q,2H,J = 7.2 Hz),2.46 (s,3H),1.01 (t,3H,J = 7.2 Hz); 13C NMR (100 MHz,CDCl3): δ 168.11,167.71,165.07,161.83,137.00, 135.66,129.16,128.41,116.09,61.17,22.67,13.53; HRMS: calcd. for C14H15ClN3O2 [M+H]+: 293.0847; found 293.0851.
Ethyl 2-amino-4-(4-bromophenyl)-6-methylpyrimidine-5-carboxylate (2d): White solid,mp 138-139 ℃. 1H NMR (400 MHz, CDCl3): δ 7.51 (d,2H,J = 8.4 Hz),7.36 (d,2H,J = 8.4 Hz),5.88 (s,2H), 4.08-4.03 (m,2H),2.43 (s,3H),0.98 (t,3H,J = 7.0 Hz); 13C NMR (100 MHz,CDCl3): δ 168.09,167.78,165.19,161.92,137.49, 131.39,129.42,123.95,116.11,61.22,22.70,13.56; HRMS: calcd. for C14H15BrN3O2 [M+H]+: 336.0342; found 336.0345.
Ethyl 2-amino-4-methyl-6-p-tolylpyrimidine-5-carboxylate (2e): White solid,mp 151-153 ℃ 1H NMR (400 MHz,CDCl3): δ 7.41 (d,2H,J = 7.6 Hz),7.20 (d,2H,J = 7.6 Hz),5.87 (s,2H),4.08 (q, 2H,J = 6.8 Hz),2.45 (s,3H),2.37 (s,3H),0.99 (t,3H,J = 7.2 Hz); 13C NMR (100 MHz,CDCl3): δ 168.62,167.23,166.30,161.96,139.60, 135.66,128.91,127.70,116.12,61.09,22.62,21.27,13.55; HRMS: calcd. for C15H18N3O2 [M+H]+: 272.1394; found 272.1400.
Ethyl 2-amino-4-(4-methoxyphenyl)-6-methylpyrimidine-5- carboxylate (2f): White solid,mp 128-130 ℃. 1H NMR (400 MHz,CDCl3): δ 7.49 (d,2H,J = 8.0 Hz),6.91 (d,2H, J = 8.0 Hz),5.95 (d,2H,J = 29.2 Hz),4.10 (q,2H,J = 7.2 Hz),3.81 (s,3H),2.42 (s,3H),1.03 (t,3H,J = 7.0 Hz); 13C NMR (100 MHz, CDCl3): δ 168.79,167.07,165.56,161.98,160.82,130.88,129.41, 115.85,113.65,61.07,55.24,22.53,13.66; HRMS: calcd. for C15H18N3O3 [M+H]+: 288.1343; found 288.1348.
Ethyl 2-amino-4-methyl-6-(4-nitrophenyl)pyrimidine-5-carboxylate (2g): White solid,mp 128-129 ℃. 1H NMR (400 MHz, CDCl3): δ 8.42 (s,1H),8.27 (d,1H,J = 8.0 Hz),7.84 (d,1H,J = 7.6 Hz), 7.58 (t,1H,J = 8.0 Hz),5.75 (s,2H),4.11 (q,2H,J = 7.2 Hz),2.49 (s, 3H),1.03 (t,3H,J = 7.2 Hz). 13C NMR (100 MHz,CDCl3): δ 168.45, 167.65,163.86,161.94,140.25,129.23,124.11,123.16,116.07, 61.43,22.98,13.64; HRMS: calcd. for C14H15N4O4 [M+H]+: 303.1088; found 303.1093.
Ethyl 2-amino-4-methyl-6-(3-nitrophenyl)pyrimidine-5-carboxylate (2h): White solid,mp 131-132 ℃. 1H NMR (400 MHz, CDCl3): δ 8.43 (s,1H),8.28 (d,1H,J = 8.0 Hz),7.85 (d,1H,J = 7.6 Hz), 7.59 (t,1H,J = 8.0 Hz),5.73 (s,2H),4.12 (q,2H,J = 6.8 Hz),2.50 (s, 3H),1.04 (t,3H,J = 7.0 Hz); 13C NMR (100 MHz,CDCl3): δ 168.45, 167.64,163.85,161.92,148.06,140.25,133.86,129.23,124.11, 123.16,116.05,61.42,22.98,13.64; HRMS: calcd. for C14H15N4O4 [M+H]+: 303.1088; found 303.1095.
Methyl 2-amino-4-(4-fluorophenyl)-6-isopropylpyrimidine-5- carboxylate (2i): White solid,mp 146-148 ℃. 1H NMR (400 MHz, CDCl3): δ 7.55 (s,2H),7.11 (t,2H,J = 6.6 Hz),5.56 (d,2H, J = 12.4 Hz),3.62 (s,3H),3.13 (s,1H),1.25 (t,6H,J = 3.2 Hz); 13C NMR (100 MHz,CDCl3): δ 175.27,169.32,164.78,164.46,162.42, 134.65,129.78 (d,J = 8.0 Hz),115.51,115.29,52.14,32.82,21.50; HRMS: calcd. for C15H17FN3O2 [M+H]+: 290.1299; found 290.1302.
6-Methyl-N2-phenylpyrimidine-2,4-diamine (2j): White solid, mp 122-124 ℃. 1H NMR (400 MHz,CDCl3): δ 7.67 (s,1H),7.52 (d, 2H,J = 7.6 Hz),7.20 (t,2H,J = 7.2 Hz),6.90 (t,1H,J = 7.4 Hz),5.70 (s, 1H),4.77 (s,2H),2.17 (s,3H); 13C NMR (100 MHz,CDCl3): δ 165.97, 163.81,159.45,139.89,128.64,121.97,119.32,95.48,23.25; HRMS: calcd. for C11H13N4 [M+H]+: 201.1135; found 201.1139.
6-Methyl-N2-o-tolylpyrimidine-2,4-diamine (2k): White solid, mp 188-190 ℃. 1H NMR (400 MHz,CDCl3): δ 7.95 (d,1H, J = 8.0 Hz),7.11 (q,2H,J = 8.0 Hz),6.90 (t,1H,J = 7.2 Hz),6.60 (s, 1H),5.71 (s,1H),4.60 (s,2H),2.21 (s,3H),2.17 (s,3H); 13C NMR (100 MHz,CDCl3): δ 166.73,163.89,160.33,137.96,130.28, 128.36,126.34,122.95,121.83,95.47,23.76,18.10; HRMS: calcd. for C12H15N4 [M+H]+: 215.1291; found 215.1295.
6-Methyl-N2-m-tolylpyrimidine-2,4-diamine (2l): Yellow oil. 1H NMR (400 MHz,CDCl3): δ 7.38-7.26 (m,3H),7.09 (t,1H, J = 7.6 Hz),6.72 (d,1H,J = 7.2 Hz),5.70 (s,1H),4.75 (s,2H),2.24 (s, 3H),2.17 (s,3H); 13C NMR (100 MHz,CDCl3): δ 166.26,163.84, 159.72,139.83,138.43,128.52,122.86,119.97,116.57,95.45, 23.48,21.51; HRMS: calcd. for C12H15N4 [M+H]+: 215.1291; found 215.1297.
6-Methyl-N2-p-tolylpyrimidine-2,4-diamine (2m): Yellow oil. 1H NMR (400 MHz,CDCl3): δ 7.69 (d,1H,J = 7.2 Hz),7.35 (d,2H, J = 7.2 Hz),6.98 (d,2H,J = 7.6 Hz),5.66 (s,1H),4.74 (s,2H),2.20 (s, 3H),2.11 (s,3H); 13C NMR (100 MHz,CDCl3): δ 166.38,163.85, 159.93,137.30,131.53,129.17,119.79,95.34,23.54,20.69; HRMS: calcd. for C12H15N4 [M+H]+: 215.1291; found 215.1294.
N2-(4-Chlorophenyl)-6-methylpyrimidine-2,4-diamine (2n): White solid,mp 136-138 ℃. 1H NMR (400 MHz,CDCl3): δ 7.72 (d,1H,J = 8.0 Hz),7.45 (d,2H,J = 8.4 Hz),7.12 (d,2H,J = 8.4 Hz), 5.72 (s,1H),4.76 (s,2H),2.15 (s,3H); 13C NMR (100 MHz,CDCl3): δ 165.48,163.74,158.89,138.43,128.45,126.60,120.41,95.62, 22.86; HRMS: calcd. for C11H12ClN4 [M+H]+: 235.0745; found 235.0758.
4-Nitroaniline: 1H NMR (CDCl3,400 MHz): δ 3.64 (s,2H),6.59- 6.61 (m,2H),7.09-7.10 (m,2H); 13C NMR (CDCl3,100 MHz): δ 116.15,123.02,129.03,144.88.
3. Results and discussionThe work was initiated with the optimization of the reaction conditions of the direct amination of pyrimidin-2-yl tosylate 1a with aqueous ammonia,utilizing 20 equiv. of sodium dodecylbenzenesulfonate (SDBS) as phase-transfer catalyst (PTC) and dioxane as solvent at 100 ℃ for 12 h (Table 1). As our prediction, the reaction afforded the amination product 2-aminopyrimidine 2a in a yield of 69% (entry 1),however,hydrolyzed product pyrimidin-2-ol 3a of 1a was also isolated in 41% yield. Lowering the temperature to 50 ℃ resulted in a higher yield of 2a (entry 2). When the SDBS was changed to hexadecyl trimethyl ammonium bromide (HTAB),cetylpyridinium chloride (CPC) and bromohexadecyl pyridine (CPB),the yield of 2a increased ([2TD$DIF]84%-89%) and trace of 3a was detected (entries 3-6). In order to find a cheaper PTC,PEG was tested. To our delight,only using PEG-200 without any other solvents,the reaction gave a good yield of 2a and the hydrolyzation of 1a was completely inhibited (entries 6 and 7). Further testing implied that PEG-400 was the best one among the PEG-200,PEG-400,PEG-600 and PEG-800 to give 2a in 86% yield (entries 7-10). Higher temperature slightly enhanced the yield at a shorter time (entries 11 and 12). Therefore,our focus was concentrated on the solvents and reaction conditions. Notably, base can greatly accelerate the translation of 1a into the byproduct 3a (82%),with the yields of 2a tremendously declined (entry 13). Download the amount of aqueous ammonia to 5 equiv. caused lower transformation (entry 14). Thus,the optimal conditions for this reaction were established: using PEG-400 as the reaction medium to perform the reaction at r.t. for 24 h.
|
|
Table 1 Optimization of conditions of pyrimidin-2-yl tosylate with NH3·H2Oa. |

{kind=link}
Under the optimized conditions,the amination of pyrimidin-2- yl tosylates (1a-i) with aqueous ammonia was tested in the reaction scope (Scheme 2). In general,good yields of the desired products were obtained. The reaction tolerated a variety of pyrimidin-2-yl tosylates containing the electron-withdrawing group as well as the electron-donating group on the phenyl ring to deliver the products (2a-i) with good yields. Compared with the previous reports,this non-catalytic approach was proven to be a powerful tool for the amines preparation in mild conditions with the lower-priced ammonia water as ammonia source [22].

|
Download:
|
| Scheme 2.Scope of the amination of pyrimidin-4-yl tosylates with NH3·H2O. | |
{kind=link}
Given the operational simplicity and broad generality of this direct amination protocol,we explored to demonstrate the utility of this strategy for the similar amine 2-aminopyrimidines using pyrimidin-4-yl tosylates (1j-n) as substrates. The desired products (2j-n) were also obtained in moderate yields under this simple reaction conditions. However,lower yields were observed,which due to the hydrolyzation of the starting materials.
To further demonstrate the versatility of the above described amination protocol,aryl- and pyridinyl tosylates were tested with aqueous ammonia. Unfortunately,only the aryl tosylate with strong electron-withdrawing substituent (NO2) underwent the amination to afford 4-nitroaniline in 50% yield. However,phenyl tosylate and pyridine-2-yl tosylate did not undergo amination with aqueous ammonia.
4. ConclusionsIn conclusion,we introduced a novel approach for amination of pyrimidinyl-2-tosylates with aqueous ammonia. The desired products 2-aminepyrimidines can be generated in high yields in mild conditions,without any catalysts or other additives. Meanwhile,the similar pyrimidin-4-yl tosylates and aryl tosylates substituted by electron-withdrawing substituents such as -NO2 afforded the desired product under the simple reaction conditions.
AcknowledgmentsFinancial support was provided by the financial support from the NSFC (Nos. 21362032 and 21362031),the Natural Science Foundation of Gansu Province (No. 1208RJYA083),Gansu Provincial Department of Finance and the Education Department of Gansu Province (No. 2013B-010).
| [1] | (a) M. Negwer, Organic Drugs and Their Synonyms, 7th ed., Akademie Verlag Gmbh, Berlin, 1994; (b) S. Suwanprasop, T. Nhujak, S. Roengsumran, A. Petsom, Petroleum marker dyes synthesized from cardanol and aniline derivatives, Ind. Eng. Chem. Res. 43 (2004) 4973-4978. |
| [2] | For reviews see: (a) D.M. Roundhill, Transition metal and enzyme catalyzed reactions involving reactions with ammonia and amines, Chem. Rev. 92 (1992) 1-27; (b) J.I. Van der Vlugt, Advances in selective activation and application of ammonia in homogeneous catalysis, Chem. Soc. Rev. 39 (2010) 2302-2322; (c) J.L. Klinkenberg, J.F. Hartwig, Catalytic organometallic reactions of ammonia, Angew. Chem. Int. Ed. 50 (2011) 86-95. |
| [3] | (a) A.W. Heinen, J.A. Peters, H. Van Bekkum, The reductive amination of benzaldehyde over Pd/C catalysts: mechanism and effect of carbon modifications on the selectivity, Eur. J. Org. Chem. 13 (2000) 2501-2506; (b) T. Gross, A.M. Seayad, M. Ahmad, M. Beller, Synthesis of primary amines: first homogeneously catalyzed reductive amination with ammonia, Org. Lett. 4 (2002) 2055-2058;(c) S. Ogo, K. Uehara, T. Abura, S. Fukuzumi, pH-dependent chemoselective synthesis of α-amino acids. Reductive amination of α-keto acids with ammonia catalyzed by acid-stable iridium hydride complexes in water, J. Am. Chem. Soc. 126 (2004) 3020-3021. |
| [4] | (a) S. Hong, T.J. Marks, Organolanthanide-catalyzed hydroamination, Acc. Chem. Res. 37 (2004) 673-686; (b) V. Lavallo, G.D. Frey, B. Donnadieu, M. Soleilhavoup, G. Bertrand, Homogeneous catalytic hydroamination of alkynes and allenes with ammonia, Angew. Chem. Int. Ed. 47 (2008) 5224-5228; (c) J. Seayad, A. Tillack, C.G. Hartung, M. Beller, Base-catalyzed hydroamination of olefins: an environmentally friendly route to amines, Adv. Synth. Catal. 344 (2002) 795-813; (d) M. Lequitte, F. Figueras, C. Moreau, S. Hub, Amination of butenes over protonic zeolites, J. Catal. 163 (1996) 255-261; (e) C.A. Tsipis, C.E. Kefalidis, How efficient are the hydrido-bridged diplatinum catalysts in the hydrosilylation, hydrocyanation, and hydroamination of alkynes: a theoretical analysis of the catalytic cycles employing electronic structure calculation methods, Organometallics 25 (2006) 1696-1706. |
| [5] | (a) E.I. du Pont de Nemours & Co., Synthesis of amines, US Patent 2497310, United States (1950).; (b) J.J. Lin, J.F. Knifton, Process for synthesis of primary amines from olefins, syngas and ammonia, US Patent 4794199, N. Texaco Inc. (White Plains), United States (1988). (c) B. Zimmermann, J. Herwig, M. Beller, The first efficient hydroaminomethylation with ammonia: with dual metal catalysts and two-phase catalysis to primary amines, Angew. Chem. Int. Ed. 38 (1999) 2372-2375. |
| [6] | (a) J. Tsuji, M. Takahashi, Palladium-catalyzed telomerization of butadiene with ammonia, J. Mol. Catal. 10 (1981) 107; (b) B. Driessen-Holscher, in: B. Cornils (Ed.), Multiphase Homogeneous Catalysis, Wiley-VCH, Weinheim, 2005, 238 and references therein. |
| [7] | (a) K. Weissermel, H.J. Arpe, Industry Organic Chemistry, Wiley-VCH, Weinheim, 1997; (b) Y.B. Jiang, W.S. Zhang, H.L. Cheng, Y.Q. Liu, R. Yang, One-pot synthesis of Naryl propargylamine from aromatic boronic acid, aqueous ammonia, and propargyl bromide under microwave-assisted conditions, Chin. Chem. Lett. 25 (2014) 779-782. |
| [8] | (a) S.A. Lawrence, Amines: Synthesis Properties, and Application, Cambridge University Press, Cambridge, 2004; (b) M.C. Willis, Palladium-catalyzed coupling of ammonia and hydroxide with aryl halides: the direct synthesis of primary anilines and phenols, Angew. Chem. Int. Ed. 46 (2007) 3402-3404; (c) S. Bahn, S. Imm, L. Neubert, et al., Synthesis of primary amines from secondary and tertiary amines: ruthenium-catalyzed amination using ammonia, Chem. Eur. J. 17 (2011) 4705-4708;(d) K. Das, R. Shibuya, Y. Nakahara, et al., Platinum-catalyzed direct amination of allylic alcohols with aqueous ammonia: selective synthesis of primary allylamines, Angew. Chem. Int. Ed. 51 (2012) 150-154. |
| [9] | Examples for palladium-catalyzed formation of aromatic amines: (a) Q. Shen, J.F. Hartwig, Palladium-catalyzed coupling of ammonia and lithium amide with aryl halides, J. Am. Chem. Soc. 128 (2006) 10028-10029; (b) D.S. Surry, S.L. Buchwald, Selective palladium-catalyzed arylation of ammonia: synthesis of anilines as well as symmetrical and unsymmetrical di-and triarylamines, J. Am. Chem. Soc. 129 (2007) 10354-10355; (c) X.H. Huang, S.L. Buchwald, New ammonia equivalents for the Pd-catalyzed amination of aryl halides, Org. Lett. 3 (2001) 3417-3419; (d) S. Lee, M. Jogensen, J.F. Hartwig, Palladium-catalyzed synthesis of arylamines from aryl halides and lithium bis(trimethylsilyl)amide as an ammonia equivalent, Org. Lett. 3 (2001) 2729-2732; (e) D.Y. Lee, J.F. Hartwig, Zinc trimethylsilylamide as a mild ammonia equivalent and base for the amination of aryl halides and triflates, Org. Lett. 7 (2005) 1169-1172; (f) X.H. Huang, K.W. Anderson, D. Zim, et al., Expanding Pd-catalyzed C-N bondforming processes: the first amidation of aryl sulfonates, aqueous amination, and complementarity with Cu-catalyzed reactions, J. Am. Chem. Soc. 125 (2003) 6653-6655; (g) J. Barluenga, F. Aznar, C. Valdes, N-trialkylsilylimines as coupling partners for Pd-catalyzed C-N bond-forming reactions: one-step synthesis of imines and azadienes from aryl and alkenyl bromides, Angew. Chem. Int. Ed. 43 (2004) 343-345; (h) J. Yin, S.L. Buchwald, Palladium-catalyzed intermolecular coupling of aryl halides and amides, Org. Lett. 2 (2000) 1101-1104. |
| [10] | Examples for copper-catalyzed formation of aromatic amines: (a) J.M. Chen, T.J. Yuan, W.Y. Hao, M.Z. Cai, Simple and efficient CuI/PEG-400 system for amination of aryl halides with aqueous ammonia, Tetrahedron Lett. 52 (2011) 3710-3713; (b) Y. Li, X.H. Zhu, F. Meng, Y.Q. Wan, Copper/oxalohydrazide/ketone catalyzed synthesis of primary arylamines via coupling of aryl halides with aqueous ammonia in water, Tetrahedron 67 (2011) 5450-5454; (c) F.Meng, X.H. Zhu, Y. Li, et al., Efficient copper-catalyzed direct amination of aryl halides using aqueous ammonia in water, Eur. J. Org. Chem. 32 (2010) 6149-6152; (d) Z.Q.Wu, Z.Q. Jiang, D.Wu, H.F. Xiang, X.G. Zhou, A simple and efficient catalytic system for coupling aryl halides with aqueousammonia in water, Eur. J. Org. Chem. 10 (2010) 1854-1857; (e) N. Xia, M. Taillefer, A very simple copper-catalyzed synthesis of anilines by employing aqueous ammonia, Angew. Chem. Int. Ed. 48 (2009) 337-339; (f) R. Ntaganda, B. Dhudshia, C.L.B. Macdonald, A. Thadani, Cross-coupling of aryl/heteroaryl bromides with ammonia using a copper-carbene catalyst, Chem. Commun. 46 (2008) 6200-6202; (g) J. Kim, S. Chang, Ammonium salts as an inexpensive and convenient nitrogen source in the Cu-catalyzed amination of aryl halides at room temperature, Chem. Commun. 26 (2008) 3052-3054; (h) F. Lang, D. Zewge, I.N. Houpis, R.P. Volante, Amination of aryl halides using copper catalysis, Tetrahedron Lett. 42 (2001) 3251-3254; (i) S. Gaillard, M.K. Elmkaddem, C. Fischmeister, C.M. Thomas, J.L. Renaud, Highly efficient and economic synthesis of new substituted amino-bispyridyl derivatives via copper and palladium catalysis, Tetrahedron Lett. 49 (2008) 3471-3474; (j) X. Gao, H. Fu, R. Qiao, Y. Jiang, Y. Zhao, Copper-catalyzed synthesis of primary arylamines via cascade reactions of aryl halideswithamidine hydrochlorides, J. Org. Chem. 73 (2008) 6864-6866; (k) H. Xu, C. Wolf, Efficient copper-catalyzed coupling of aryl chlorides, bromides and iodides with aqueous ammonia, Chem. Commun. 48 (2009) 3035-3037; (l) D.P.Wang, Q. Cai,K. Ding,Anefficient copper-catalyzedaminationof aryl halides by aqueous ammonia, Adv. Synth. Catal. 351 (2009) 1722-1726; (m) P.J. Ji, J.H. Atherton, I. Michael, Copper(I)-catalyzed amination of aryl halides in liquid ammonia, J. Org. Chem. 77 (2012) 7471-7478; (n) J.X. Zhang, H.Q. Yin, S.Q. Han, Copper-catalyzed N-arylations of nitrogen-containing heterocycles in water, Chin. J. Org. Chem. 32 (2012) 1429-1433; (o) W. Liu, Y.L. Bi, Progress in copper-catalyzed direct arylation of aromatic C-H bonds, Chin. J. Org. Chem. 32 (2012) 1041-1050. |
| [11] | C.O. Kappe, Biologically active dihydropyrimidones of the Biginelli-type-A literature survey, Eur. J. Med. Chem. 35 (2000) 1043-1052. |
| [12] | (a) C.O. Kappe, 100 years of the Biginelli dihydropyrimidine synthesis, Tetrahedron 49 (1993) 6937-6963; (b) C.O. Kappe, Recent advances in the Biginelli dihydropyrimidine synthesis. New tricks from an old dog, Acc. Chem. Res. 33 (2000) 879-888; (c) C.O. Kappe, A. Stadler, The Biginelli dihydropyrimidine synthesis, Org. React. 63 (2004) 1-117; (d) K. Singh, D. Arora, K. Singh, S. Singh, Genesis of dihydropyrimidinone calcium channel blockers: recent progress in structure-activity relationships and other effects, Med. Chem. 9 (2009) 95-106. |
| [13] | (a) Z.J. Quan, H.D. Xia, Z. Zhang, Y.X. Da, X.C. Wang, An efficient copper-catalyzed N-arylation of amides: synthesis of N-arylacrylamides and 4-amido-N-phenylbenzamides, Tetrahedron 69 (2013) 8368-8374; (b) Z.J. Quan, H.D. Xia, Z. Zhang, Y.X. Da, X.C. Wang, Copper-catalyzed amination of aryl halides with aqueous ammonia under mild conditions, Chin. J. Chem. 31 (2013) 501-506; (c) Z.J. Quan, W.H. Hu, X.D. Jia, et al., A domino desulfitative coupling/acylation/hydration process cocatalyzed by copper(i) and palladium(ii): synthesis of highly substituted and functionalized fyrimidines, Adv. Synth. Catal. 354 (2012) 2939-2948; (d) Z.J. Quan, Y. Lv, Z.J. Wang, et al., Molecular iodine-mediated S-N and C-N cross-coupling and oxidative aromatization of 3,4-dihydropyrimidin-2(1H)-thiones with secondary amines, Tetrahedron Lett. 54 (2013) 1884-1887; (e) Z.J. Quan, W.H. Hu, Z. Zhang, et al., One-pot synthesis of allylamine derivatives by iodine-catalyzed three-component reaction of N-heterocycles, paraformaldehyde and styrenes, Adv. Synth. Catal. 355 (2013) 891-900; (f) Y.X. Da, Z. Zhang, Z.J. Quan, Intermolecular cyclocondensation reaction of 3,4-dihydropyrimidine-2-thione under the Mitsunobu reaction conditions, Chin. Chem. Lett. 22 (2011) 679; (g) X.C. Wang, G.J. Yang, Z.J. Quan, P.Y. Ji, J.L. Liang, R.G. Ren, Synthesis of 2-substituted pyrimidines via cross-coupling reaction of pyrimidin-2-yl sulfonates with nucleophiles in polyethylene glycol 400, Synlett 11 (2010) 1657-1660. |
| [14] | For a review, see: I.M. Lagoja, Pyrimidine as constituent of natural biologically active compounds, Chem. Biodiversity 2 (2005) 1-50. |
| [15] | (a) K.B. Goodman, D. Lee, C.A. Sehon, A.Q. Viet, G.Z. Wang, Novel inhibitors of rhokinases, Int. Patent Appl. WO 2006009889 (2006).; (b) D. Drewry, B. Evans, K.B. Goodman, et al., Chemical compounds, Int. Patent Appl. WO 2004112719 A8 (2004). |
| [16] | J.M. Nuss, S.D. Harrison, D.B. Ring, et al., U.S. Patent 6,417,185 (2002); Chem. Abstr. 137 (2002) 325431 |
| [17] | S. Fujita, M. Hagihara, S. Iwayama, et al., Novel pyrimidine derivative and novel pyridine derivative, Int. Patent Appl. WO 2002022588 A1 (2002) |
| [18] | M. Watanabe, H. Koike, T. Ishiba, et al., Synthesis and biological activity of methanesulfonamide pyrimidine-and N-methanesulfonyl pyrrole-substituted 3,5-dihydroxy-6-heptenoates, a novel series of HMG-CoA reductase inhibitors, Bioorg. Med. Chem. 5 (1997) 437-444. |
| [19] | Some examples for the synthesis of Rosuvastatin involves 2-amino-pyrimidine-5-carboxylates, see:; (a) V. Niddam-Hildesheim, K. Chen, A process for the preparation of rosuvastatin involving a tempo-mediated oxidation step, Int. Patent Appl. WO 2006017357 (2006).; (b) S. Gudipati, S. Katkam, R.R. Sagyam, J.S. Kudavalli, Processes to produce intermediates for rosuvastatin, U.S. Patent 2006004200 (2006).; (c) S. Ahmad, J.A. Robl, K. Ngu, Pyrimidine and pyridine derivatives useful as hmgcoa reductase inhibitors and method of preparation thereof, Int. Patent Appl. WO 2005030758 (2005).; (d) N. End, Y. Richter, Process for the preparation of pyrimidine derivatives, Int. Patent Appl. WO 2004103977 (2004). |
| [20] | R. Capdeville, E. Buchdunger, J. Zimmermann, A. Matter, Glivec (ST1571, Imatinib), a rationally developed, targeted anticancer drug, Nat. Rev. Drug Discov. 1 (2002) 493-502. |
| [21] | (a) P. Dorigo, D. Fraccarollo, G. Santostasi, et al., Synthesis and cardiotonic activity of novel pyrimidine derivatives: crystallographic and quantum chemical studies, J. Med. Chem. 39 (1996) 3671-3683; (b) S. Nagarajan, P. Shanmugavelan, M. Sathishkumar, et al., An eco-friendly water mediated synthesis of 1,2,3-triazolyl-2-aminopyrimidine hybrids as highly potent anti-bacterial agents, Chin. Chem. Lett. 25 (2014) 419-422. |
| [22] | M. Matloobi, C.O. Kappe, Microwave-assisted solution-and solid-phase synthesis of 2-amino-4-arylpyrimidine derivatives, ACS. Comb. Sci. 9 (2007) 275-284. |