




The incorporation of a trifluoromethyl group into a molecule usually modifies its chemical and physical properties through steric and electronic effects [1]. As a result,intensive research efforts have been directed toward the development of efficient methods for trifluoromethylation [2]. In the recent years,transition metals have proved to be effective for promoting trifluoromethylation because of their high level of functional group tolerance and mild reaction conditions [3]. Cu and its salts are the most-widely used transition metal in trifluoromethylation because of their low cost and good reactivity. In the reaction mechanism studies of Cu-promoted trifluoromethylation,trifluoromethyl copper ‘‘CuCF3’’ has been proposed as an important intermediate [4]. However,in most of cases,the structure of ‘‘CuCF3’’ is elusive and complex,which makes the understanding and exploration of reaction mechanism very difficult [5]. Very recently,Grushinet al. [6] reported their computational study of the trifluoromethylation reaction of aryl halides with fluoroform-derived CuCF3,which is a ‘‘ligandless’’ CuCF3with weak coordinating ligand such as DMF, ET3 Nortert-BuOH. However,the mechanism of adequately characterized CuCF3 complexes has not been researched by computational methods. In 2008,the first examples of isolable and structurally characterized CuI -CF3 complexes are reported by Vicicet al. [7]. N-Heterocyclic carbene (NHC)-supported copper tert-butoxide complexes reacted with Me3 Si-CF3 to afford new (NHC)Cu-CF3 complexes (1 and 2in Scheme 1). In situ generated well-defined (SIiPr)Cu-CF3 (1) cleanly transferred its trifluoromethyl group to a number of organic halides under mild conditions. Based on this discovery,we calculate the reaction pathways from the metal complexes to the final benzotrifluoride by the use of density functional theory. It is helpful to understand the mechanism of trifluoromethylation reactions of iodobenzene with well-defined NHC copper trifluoromethyl complexes.

|
Download:
|
| Scheme 1.Computational models of reagents employed in trifluoromethylation reactions of aryl halides. | |
{kind=link}
In order to gain a deeper insight into the mechanism of the reaction of (NHC)CuCF3 with aryl halides. Density functional theory (DFT) calculations based on the M06 level [8],and the 6-31+G* basis set [9] was employed for the C,H,N,F and Si atoms,the LANL2DZ effective core potentials of Hay and Wadt with double-z basis sets [10] for copper and iodine atoms. The DFT calculations were carried out using the Gaussian 09 programs [11]. All geometries were fully optimized without any constraints in the N,N-dimethylformamide (DMF) with SMD [12] model was applied for solvent effect. Intrinsic reaction coordinate (IRC) calculations were carried out to confirm the connectivities between the minima and transition states involved in the oxidative addition steps. In this article,all energies correspond to the computed Gibbs free energies in DMF. The vibrational frequency was computed for all structures in order to determine whether it is an intermediate (no imaginary frequency) or transition state (one and only one imaginary frequency). 3. Results and discussion
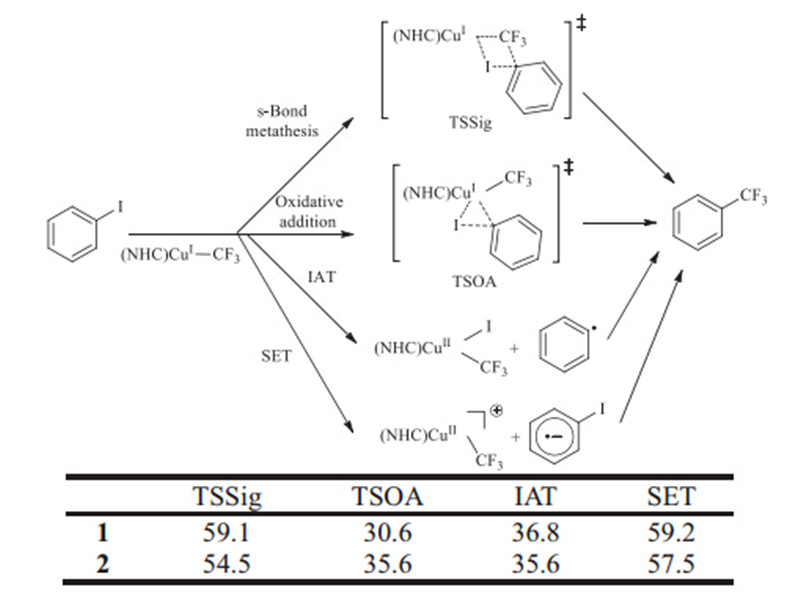
Vicic reported that copper trifluoromethyl complex 1 was generated by using two equivalents of CF3 SiMe3 /KF with a saturatedN-heterocyclic carbene-supported coppertert-butoxide, and reaction ofin situgenerated1with 5 equivalents of aryl halides gave high yields (91-99%) of trifluoromethylated arenes in DMF after 112 h. Similarly,complex 1 was synthesized and reaction of2 with Ph-I at room temperature for 44 h led to Ph-CF3 in 33% yield [7]. Our efforts have been aimed to elucidate the mechanism for the trifluoromethylation of iodobenzene with well-defined trifluoromethyl copper complexes as shown in Scheme 1. By using computational models of the reagents used in the experiments,we can reduce the computational cost. We propose that the reaction may proceedvia four reaction pathways,such as s-bond metathesis (BM),concerted oxidative addition-reductive elimination (OARE),iodine atom transfer (IAT) and single-electron transfer (SET). These four reaction pathways were often seen in reports of a broad variety of Cu-promoted aromatic coupling reactions [13].
Computed free energies for key species in possible mechanisms for iodobenzene activation of copper complexes 1 and 2 are showed in Scheme 2. The energies computed for key complexes in the single-electron transfer mechanism are much larger than the activation energies for both oxidation of iodobenzene to the two CuI CF3 complexes. The energies for key species of the SET pathways for 1 and 2are 59 and 57 kcal/mol respectively. Additionally, frontier molecular orbital (FMO) analysis found that the lowest unoccupied molecular orbital (LUMO) of iodobenzene is -0.62 eV and the highest occupied molecular orbitals (HOMO) of1 and 2 were -6.30 and -6.26 eV,which was too low to take place an electron transfer from1 and 2to iodobenzene. The activation energies fors-bond metathesis (BM) are also prohibitively large and almost isoenergetic with the free energies of formations of SET intermediates. The energies required for s-bond metathesis of iodobenzene with1 and 2are 59 and 55 kcal/mol,respectively. The unreasonable high barriers suggest that both SET and BM pathways for the trifluoromethylation by the well-defined complexes1 and 2are unfavorable.
|
|
Download:
|
| Scheme 2.Free energies (kcal/mol) for key stationary points in the mechanism of trifluoromethylation reactions of iodobenzene with well-defined copper complexes 1 and 2. | |
{kind=link}
The energy computed for key complexes in iodine atom transfer to1is 6 kcal/mol,higher than the activation energy for oxidation addition of iodobenzene to CuI CF3 complex 1. Although the energy computed for a key complex in iodine atom transfer for complex 2 is close to the oxidation addition barrier of iodobenzene to complex 2,a higher barrier is required for IAT on the basis of Marcus-Hush theory [14]. The oxidation addition mechanisms for complexes1 and 2therefore are favored.
The barrier for oxidation addition of Ph-I to well-defined complex 1 is 31 kcal/mol. Very recently,Grushin and coworkers have studied the trifluoromethylation reaction of a variety of aryl halides with fluoroform-derived CuCF3 by experimental and computational means. In their study,the computed barrier for the trifluoromethylation of iodobenzene involving Ph-I oxidation to DMF stabilized CuCF3is 22 kcal/mol,which is in good agreement with the experimental value of 24 kcal/mol [6]. They also shows that the replacement of DMF in [(DMF)CuCF3] with NMe3 raises the activation barrier by 2.4 kcal/mol and NMe3 binds to CuCF3 7 kcal/mol more strongly than DMF. Their calculation implies that the reactivity of trifluoromethylation decreases with the increase of the binding ability of ligand. Our calculation shows that the free energy of NHC ligated CuCF3(1) is about 22 kcal/mol lower than that ligated by DMF (see Supporting information for detail). This high energy is in agreement with a high energy of TSOA for1and also in agreement with the slow reaction rate observed by the experiments [7].
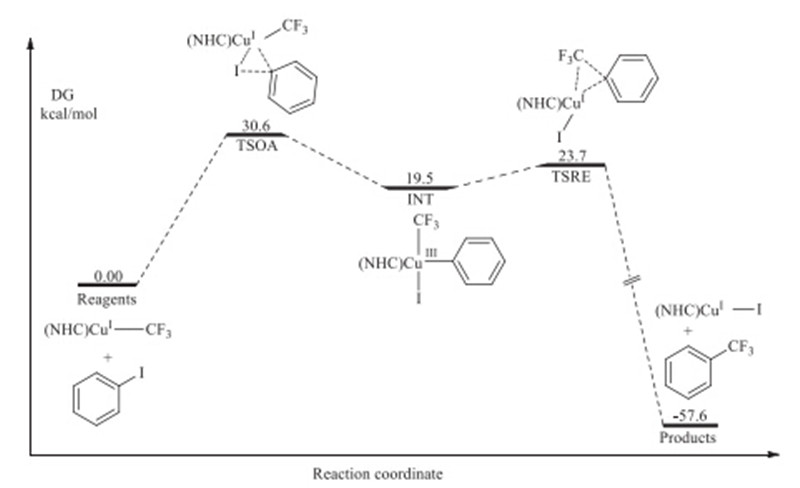
The detailed profile of the oxidative addition-reductive elimination mechanism for 1 is shown in Fig. 1. TSOA with the highest energy suggests that the oxidation addition of Ph-I to NHC-ligated Cu complex is the rate determining step. The generation of intermediate (INT) from TSOA with high energy is exergonic,resulting a relatively low energy of CuIII complex. The computed structure of INT shows that CuIII center of the intermediate is planar quadrilateral. The transition structure of reductive elimination (TSRE) is only 4 kcal/mol higher than INT,so that this step is easy to occur. The calculated overall free energy effect of -58 kcal/mol shows that the trifluoromethylation process is highly exothermic.

|
Download:
|
| Fig. 1. Free-energy profile of concerted OARE trifluoromethylation reaction of iodobenzene with well-defined copper complex 1. | |
{kind=link}
Free-energy profile of concerted OARE trifluoromethylation reaction of iodobenzene with well-defined copper complex 2,freeenergies of ligand exchange between DMF and 1 or 2 and calculated imaginary frequencies of transition states are provided in the Supporting information. Ball-and-stick model structure, structure parameters (Å or˚),computed energies (a.u.),and cartesian coordinates (Å ) for all transition states and intermediates,and the estimated barriers for SET and IAT were calculated with Marcus-Hush theory,and the intrinsic reaction coordinate (IRC) calculation was performed for two TSOA involved in the reactions. 4. Conclusion
In summary,a DFT computational study to investigate the mechanism of trifluoromethylation reaction of iodobenzene with well-defined (NHC)CuCF3complexes has been accomplished. The barriers of oxidation addition-reductive elimination ands-bond metathesis mechanisms,as well as free energies for key species in single electron transfer and iodine atom transfer pathways,are computed. Although the barrier is higher than that for (DMF)CuCF3,the concerted OARE pathway is favored with the rate determining step being the oxidation addition of Ph-I to (NHC)CuCF3complex and is highly exothermic. The current results are consistent with the mechanism study of the trifluoromethylation reaction with fluoroform-derived CuCF3[6].
AcknowledgmentsThis work was supported by National Natural Science Foundation of China (Nos. 21073144,21173169) and the Fundamental Research Funds for the Central Universities (No. XDJK2013A008). Computing resources were provided by the National Supercomputing Center of China in Shenzhen.
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet. 2014.12.017.
| [1] | (a) D. O’Hagan, Understanding organofluorine chemistry. An introduction to the C-F bond, Chem. Soc. Rev. 37 (2008) 308-319;(b) K. Mü ller, C. Faeh, F. Diederich, Fluorine in pharmaceuticals: looking beyond intuition, Science 317 (2007) 1881-1886; (c) J. Wang, M. Sánchez-Roselló, J.L. Aceña, et al., Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade(2001-2011), Chem. Rev. 114 (2014) 2432-2506; (d) S. Purser, P.R. Moore, S. Swallow, V. Gouverneur, Fluorine in medicinalchemistry, Chem. Soc. Rev. 37 (2008) 320-330; (e) C.H. Ge, R. Zhang, P. Fan, et al., Supramolecular assembly of 2,4,5-trifluorobenzoatecomplex based on weak interactions involving fluorine atoms, Chin.Chem. Lett. 24 (2013) 73-75; (f) X.J. Song, P. Yang, H. Gao, et al., Facile synthesis and antitumor activity of novel2-trifluoromethylthieno[2,3-d]pyrimidine derivatives, Chin. Chem. Lett. 25(2014) 1006-1010. |
| [2] | (a) F. Qing, Recent advances of trifluoromethylation, Chin. J. Org. Chem. 32 (2012)815-824;(b) J.A. Ma, D. Cahard, Asymmetric fluorination, trifluoromethylation, and perfluoroalkylationreactions, Chem. Rev. 108 (2008) PR1-PR43; (c) J. Nie, H.C. Guo, D. Cahard, J.A. Ma, Asymmetric construction of stereogeniccarbon centers featuring a trifluoromethyl group from prochiral trifluoromethylatedsubstrates, Chem. Rev. 111 (2010) 455-529; (d) S. Barata-Vallejo, A. Postigo, Metal-mediated radical perfluoroalkylation oforganic compounds, Coord. Chem. Rev. 257 (2013) 3051-3069. |
| [3] | (a) O.A. Tomashenko, V.V. Grushin, Aromatic trifluoromethylation with metalcomplexes, Chem. Rev. 111 (2011) 4475-4521;(b) S. Roy, B.T. Gregg, G.W. Gribble, V.D. Le, S. Roy, Trifluoromethylation of aryland heteroaryl halides, Tetrahedron 67 (2011) 2161-2195; (c) C.P. Zhang, Q.Y. Chen, Y. Guo, J.C. Xiao, Y.C. Gu, Difluoromethylation andtrifluoromethylation reagents derived from tetrafluoroethane β-sultone: synthesis,reactivity and applications, Coord. Chem. Rev. 261 (2014) 28-72; (d) T. Liang, C.N. Neumann, T. Ritter, Introduction of fluorine and fluorine-containingfunctional groups, Angew. Chem. Int. Ed. 52 (2013) 8214-8264. |
| [4] | (a) V.C.R. McLoughlin, J. Thrower, A route to fluoroalkyl-substituted aromaticcompounds involving fluoroalkylcopper intermediates, Tetrahedron 25 (1969)5921-5940;(b) Q.Y. Chen, S.W. Wu, Methyl fluorosulphonyldifluoroacetate; a new trifluoromethylatingagent, J. Chem. Soc., Chem. Commun. (1989) 705-706; (c) M. Oishi, H. Kondo, H. Amii, Aromatic trifluoromethylation catalytic in copper,Chem. Commun. (2009) 1909-1911. |
| [5] | D.M. Wiemers, D.J. Burton, Pregeneration, spectroscopic detection and chemicalreactivity of (trifluoromethyl)copper, an elusive and complex species, J. Am.Chem. Soc. 108 (1986) 832-834. |
| [6] | A.I. Konovalov, A. Lishchynskyi, V.V. Grushin, Mechanism of trifluoromethylationof aryl halides with CuCF3 and the ortho effect, J. Am. Chem. Soc. 136 (2014)13410-13425. |
| [7] | G.G. Dubinina, H. Furutachi, D.A. Vicic, Active trifluoromethylating agents fromwell-defined copper(I)-CF3 complexes, J. Am. Chem. Soc. 130 (2008) 8600-8601. |
| [8] | Y. Zhao, D.G. Truhlar, A new local density functional for main-group thermochemistry,transition metal bonding, thermochemical kinetics, and noncovalentinteractions, J. Chem. Phys. 125 (2006) 194101. |
| [9] | (a) W.J. Hehre, R. Ditchfield, J.A. Pople, Self-consistent molecular orbital methods.XII. Further extensions of Gaussian-type basis sets for use in molecular orbitalstudies of organic molecules, J. Chem. Phys. 56 (1972) 2257-2261;(b) P.C. Hariharan, J.A. Pople, The influence of polarization functions on molecularorbital hydrogenation energies, Theor. Chim. Acta 28 (1973) 213-222; (c) M.M. Francl, W.J. Pietro, W.J. Hehre, et al., Self-consistent molecular orbitalmethods. XXIII. A polarization-type basis set for second-row elements, J. Chem.Phys. 77 (1982) 3654-3665. |
| [10] | (a) P.J. Hay, W.R. Wadt, Ab initio effective core potentials for molecular calculations.Potentials for K to Au including the outermost core orbitals, J. Chem. Phys.82 (1985) 299-310;(b) W.R. Wadt, P.J. Hay, Ab initio effective core potentials for molecular calculations.Potentials for main group elements Na to Bi, J. Chem. Phys. 82 (1985) 284-298; (c) P.J. Hay, W.R. Wadt, Ab initio effective core potentials for molecular calculations.Potentials for the transition metal atoms Sc to Hg, J. Chem. Phys. 82 (1985)270-283. |
| [11] | M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 09, Revision D, Gaussian,Inc., Wallingford, CT, 2013. |
| [12] | A.V. Marenich, C.J. Cramer, D.G. Truhlar, Universal solvation model based onsolute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensions, J. Phys. Chem. B 113 (2009)6378-6396. |
| [13] | (a) G. Lefèvre, G. Franc, A. Tlili, et al., Contribution to the mechanism of coppercatalyzedC-N and C-O bond formation, Organometallics 31 (2012) 7694-7707; (b) I.P. Beletskaya, A.V. Cheprakov, The complementary competitors: palladiumand copper in C-N cross-coupling reactions, Organometallics 31 (2012) 7753-7808; (c) G.O. Jones, P. Liu, K.N. Houk, S.L. Buchwald, Computational explorations ofmechanisms and ligand-directed selectivities of copper-catalyzed Ullmann-typereactions, J. Am. Chem. Soc. 132 (2010) 6205-6213; (d) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz, M. Lemaire, Aryl-aryl bondformation one century after the discovery of the Ullmann reaction, Chem. Rev.102 (2002) 1359-1470; (e) Z. Weng, W. He, C. Chen, et al., An air-stable copper reagent for nucleophilictrifluoromethylthiolation of aryl halides, Angew. Chem. Int. Ed. 52 (2013) 1548-1552. |
| [14] | (a) A. Houmam, Electron transfer initiated reactions: bond formation and bonddissociation, Chem. Rev. 108 (2008) 2180-2237;(b) C.Y. Lin, M.L. Coote, A. Gennaro, K. Matyjaszewski, Ab initio evaluation of thethermodynamic and electrochemical properties of alkyl halides and radicals andtheir mechanistic implications for atom transfer radical polymerization, J. Am.Chem. Soc. 130 (2008) 12762-12774. |