



 , Herman H.Y. Sungb, Ian D. Williamsb, Chi K. Changb,c
, Herman H.Y. Sungb, Ian D. Williamsb, Chi K. Changb,c
b Department of Chemistry, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China;
c Department of Chemistry, Michigan State University, East Lansing, MI 48824, USA
Porphyrin and its analogues play an important role in a wide variety of areas such as luminescent dyes [1, 2, 3],electron transfer [4] and catalysis [5]. Halogenated porphyrins are of great interests for their unique catalytic [6, 7] and nonlinear optical properties [8]. Halogen atom may either directly attached at meso-carbons [9] or at β-pyrrole positions [10, 11] of porphyrin macrocycle. The convenient halogenation method for porphyrins is the mesosubstitution by using halogenated aryl groups [12]. It is well known that β-halogenation has remarkable influence on the properties of porphyrins and metalloporphyrins. For example,bbromination and β-chlorination will cause red-shift in the UV-vis spectra of porphyrins,while β-fluorination will bring a large blueshift in its UV-vis spectrum [13]. Generally,β-fluorinated porphyrins have quasiplanar macrocyclic structure,upfieldshifted NH resonances in the 1H NMR spectra,and lower energy levels of both HOMOs and LUMOs as well as higher efficiency in catalytic oxidation reactions [14]. So far,β-octafluoroporphyrins [14] and β-octafluorocorroles [15, 16] have to employ the condensation of 3,4-difluoropyrrole [17] and aldehyde in the presence of acid catalyst.
Meso-tetraalkyl porphyrins represent a kind of porphyrins bearing four alkyl groups on their meso-positions. Mesosubstitution has significant effects on the reactivity of porphyrin macrocycle,especially the reactions involving free radicals [18, 19]. Meso-tetraalkyl porphyrins serve as useful probes for understanding the structure-property relationship of porphyrin molecules [20]. Furthermore,meso-tetraalkyl glyoxylate ester porphyrin is found to be a powerful precursor for the preparation of porphine [21],which is obtained in a very low yield by traditional one-pot Rothemund pyrrole-formaldehyde condensation method [22]. Metal meso-tetra(alkoxycarbonyl)- porphyrin exhibits superior catalase-like activity as compared to its tetraphenylporphyrin counterparts [23]. In addition, tetrakis(alkoxycarbonyl)porphyrin is a good precursor for the synthesis of tetracarboxylporphyrin with the carboxyl groups directly linked at meso-positions. This may constitute a new class of building blocks for the construction of carboxylporphyrin metal organic frameworks [24]. The only report that exists in literature about the synthesis of tetra(alkoxycarbonyl) porphyrin is the condensation of glyoxylate esters and pyrrole in the presence of BF3·OEt2. This reaction was carried out in large scale (up to 19 L solution),probably to avoid scrambling to generate the final porphyrin product [23]. Less reports on application studies of these porphyrin macrocycles may be partially ascribed to the lack of further synthetic improvements of them. In connection to our efforts to synthesize new porphyrinoids for use in catalysis,we herein wish to report the convenient synthesis of a novel highly electron-deficient β-octafluoro-meso-tetra(alkoxycarbonyl)- porphyrin: 2,3,7,8,12,13,17,18-Octafluoro-5,10,15,20-tetrakis( ethoxycarbonyl)porphyrin 2 (Scheme 1). The synthetic reaction was carried out by using condensation of 3,4- difluoro-1H-pyrrole and ethyl glyoxylate in 80 mL of dichloromethane solution,giving an isolated yield of~8%. 5,10,15,20- tetrakis(ethoxycarbonyl)porphyrin 1 was also prepared by this modified method with almost similar yield. Single crystal structure of corresponding zinc complexes showed that both Zn-1 and Zn-2 have planar molecular structure having six coordinated central zinc atom.

|
Download:
|
| Scheme 1. Molecular structures of porphyrins and their zinc(II) complexes. | |
All reagents were purchased from commercial sources and used without further purification. UV-vis absorption spectra were recorded on Hitachi 3900H UV-vis spectrophotometer in quartz cell with path length of 1 cm at room temperature,and emission spectra were recorded on PerkinElmer LS55 fluorescence spectrophotometer. HR-MS spectra were recorded on Bruker maxis impact mass spectrometer with an ESI source. 1H NMR and 19F NMR spectra were recorded on Bruker Avance III 400 MHz spectrometer in CDCl3 or DMSO-d6 solution. Single-crystal X-ray diffraction data were recorded on a Rigaku R-AXIS SPIDER IP diffractometer with Mo Kα radiation. 2.1. Synthesis of free base porphyrins
5,10,15,20-Tetrakis(ethoxycarbonyl)porphyrin (1): Under nitrogen atmosphere,1.33 mL of ethyl glyoxylate in toluene solution (50%,6.5 mmol) and 0.468 mL of freshly distilled pyrrole (6.7 mmol) were dissolved in dichloromethane (500 mL) in a 1 L round-bottom flask equipped with a magnetic stirrer. The mixture was degassed for 10 min,followed by injection of boron trifluoride dietherate (BF3·Et2O) 0.2 mL (46.8%-47.8%,~1.6 mmol) into the reaction mixture. After stirring for 100 min at room temperature, the reaction was quenched by triethylamine (1.0 mL). 1.48 g (6.5 mmol) 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) was introduced and the stirring was continued for another 1 h. The crude reaction mixture was evaporated to dryness and the residue was purified by silica-gel column chromatography using dichloromethane/hexane (4:1,v/v) as eluent. The pure product was obtained after recrystallization from dichloromethane/hexane (1:4 v/v). Yield: 100 mg (10%). MS (HR-MS) calcd. for C32H30N4O8:598.2064; found: m/z 599.2142 [M + 1]+; 1H NMR (400 MHz, CDCl3) δ 9.54 (s,8H),5.12 (q,8H,J = 7.1 Hz),1.82 (t,12H,J = 7.1 Hz), -3.35 (s,2H); 13C NMR (100 MHz,CDCl3,TMS) δ 170.48,145.38, 131.43,112.18,63.49,14.77; UV-vis (CH2Cl2) λmax (log ε) 407 (5.33),505 (4.20),540 (3.59),583 (3.75),638 (3.44).
2,3,7,8,12,13,17,18-Octafluoro-5,10,15,20-tetrakis(ethoxycarbonyl) porphyrin (2): Under nitrogen atmosphere,ethyl glyoxylate in toluene solution 333 μL (50%,1.6 mmol) and 3,4-difluoropyrrole 174 mg (1.7 mmol) were dissolved in dichloromethane (80 mL) in a 150 mL round-bottom flask equipped with a magnetic stirrer. The mixture was degassed for 10 min followed by injection of BF3·Et2O 0.05 mL (46.8-47.8%,0.4 mmol) into the reaction mixture. The reaction mixture was stirred at room temperature for 100 min. Triethylamine (0.3 mL) was added to quench the reaction. 0.5 g (2.2 mmol) 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) was introduced to the reaction mixture and the stirring was continued for a further 1 h. The reaction mixture was filtrated on a short silica gel column,and then the solvent was removed by rotary evaporation under vacuum to afford crude product. The crude product was washed with hexane and ethyl ether successively. The pure product was obtained after recrystallization from dichloromethane/hexane (1:4 v/v). Yield: 60 mg (8%). MS (HR-MS) calcd. for C32H22F8N4O8: 742.1310; found: m/z 743.1377 [M + 1]+; 1H NMR (400 MHz,CDCl3) δ 5.13 (q,8H,J = 7.1 Hz),1.84 (t,12H,J = 7.1 Hz),-7.35 (s,2H); UV-vis (CH2Cl2) λmax (log ε) 385 (5.16),486 (4.22),523 (3.27),572 (3.68),625 (3.09). 2.2. Synthesis of Zn-1 and Zn-2
1 or 2 (30 mg) was dissolved in DMF (10 mL) and stirred for about 1 h along with the excess amounts of zinc acetate. After concentration,column chromatography (silica gel,dichloromethane) was carried out to remove free-base porphyrin. Further elution with dichloromethane/methanol (99:1 v/v) afforded Zn-1 or Zn-2,Yield: 98%. Zn-1: UV-vis. (CH2Cl2) λmax 412 (5.59),546 (4.26),582 (3.43); 1H NMR (400 MHz,CDCl3) δ 9.56 (s,8H),5.09 (q, 8H,J = 7.1 Hz),1.74 (t,13H,J = 7.1 Hz); MS (HR-MS) calcd. for C32H28N4O8Zn: 660.1199; found: m/z 661.1268 [M + 1]+. Zn-2: UV- vis (CH2Cl2) λmax 402 (5.03),535 (3.85),567 (2.85); 1H NMR (400 MHz,CDCl3) δ 4.98 (dd,8H,J = 13.4,6.5 Hz),1.71 (t,12H, J = 7.5 Hz); MS (HR-MS) calcd. for C32H20F8N4O8Zn: 804.0445; found: m/z 805.0510 [M + 1]+. 2.3. X-ray diffraction studies of Zn-1 and Zn-2
Crystallographic data for Zn-1 and Zn-2 was collected at 100(2) K using Mo Kα radiation (λ = 0.71073Å ). The data was corrected for Lorentz and polarization factors and for absorption by using Semi-empirical from equivalents scan data. The structure was solved with the SHELX program,and refined by full-matrix leastsquares method based on F2,with anisotropic thermal parameters for the non-hydrogen atoms. The hydrogen atoms were generated by the riding mode. Data collection and structural refinement parameters are given in Table 1. Selected bond distances and bond angles for Zn-1 and Zn-2 are shown in Table S1 and S2 (see Supporting information). CCDC 1025286 and 1025285 contained the supplementary crystallographic data for Zn-1 and Zn-2.
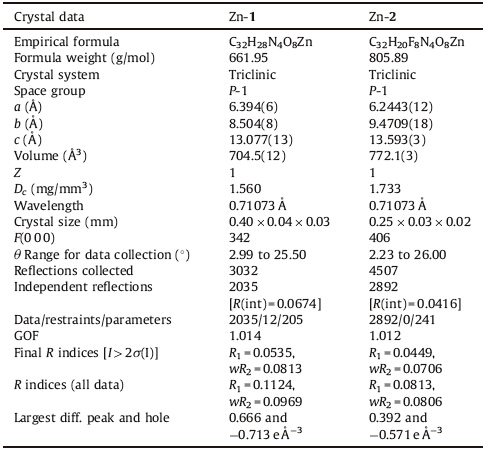
| Table 1 Crystal structure data and refinement for Zn-1 and Zn-2. |
β-Octafluorinated tetrakis(ethoxycarbonyl)porphyrin (2) was prepared by the acid-catalyzed condensation of 3,4-difluoropyrrole and the corresponding ethyl glyoxylate in 80 mL dichloromethane solution,followed by oxidation with DDQ. The synthetic procedures generally followed those outlined by Lindsey [25], except that a greater than stoichiometric amount of boron trifluoride dietherate was employed and the reaction was quenched with triethylamine after only 100 min. The isolated yield is~8%,similar to the unfluorinated porphyrin 1,albeit its solubility in dichloromethane is poor at room temperature.
The absorption spectra of 1 and 2 (Fig. S1 in Supporting information) showed large blue shifts in comparison to mesotetrakis( 4-methoxycarbonylphenyl)porphyrin (TMeCPP),particularly in the Soret (B) band region of the spectra (Table 2). The energy difference B (0,0)-Q (0,0) for synthesized porphyrins is also given in Table 2. The electronic interactions between the two pairs of nearly degenerate lowest excited singlet give the B and Q bands [26, 27]. The extent of the electronic interactions and,thus,the magnitude of the B (0,0)-Q (0,0) splitting is affected by two main factors: the degeneracy of the states and the compactness of the orbitals involved. When the frontier orbitals are delocalized onto peripheral substituents,the overall electronic interactions are smaller. Compared with meso-tetraarylporphyrins,porphyrin 1 and 2 have better planarity and smaller conjugated,which results in 1 and 2 possess extremely large B (0,0)-Q (0,0) energy differences. A remarkable feature that appears in the electronic absorption spectra of β-octafluorinated porphyrin 2 and Zn-2 is a clear hypsochromic shift compared with the unfluorinated counterparts (Table 2). For example,the Soret absorption band of 2 was found at 385 nm (cf. 407 nm for 1),while that of Zn-2 was found at 402 nm (cf. 412 nm for Zn-1). Compounds 2 and Zn-2 also displayed a larger splitting between the B (0,0) and Q (0,0) band and a small molar extinction coefficient for Q (0,0),similar to the previous observations [27, 28, 29],and this can be interpreted as a direct effect of the electronegativity of fluorine. It is noteworthy that the fluorescence of β-octaflurinated porphyrin 2 and Zn-2 in dichloromethane are significantly weaker than corresponding unfluorinated compounds,(Fig. S2 in Supporting information),and this may be due to the higher aggregation tendency of 2 and Zn-2.

|
Download:
|
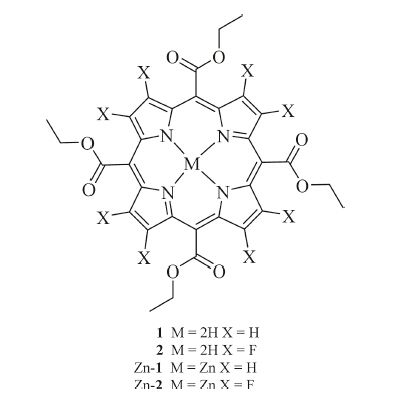
| Fig 1. ORTEP drawing of Zn-1 and Zn-2. | |
| Table 2 Spectroscopic data of synthesized porphyrins. |

{kind=link}
{kind=link}
The 1H NMR spectrum of the free-base β-fluorinated porphyrin 2 displays upfield-shifted NH protons (-7.35 ppm) as compared to the unfluorinated porphyrin (-3.35 ppm) (Table 2). The chemical shift for NH protons in compound 1 is comparable to that in boctamethylporphyrin (-3.76 ppm) but still more negative than TMeCPP (-2.78 ppm),typical of symmetrical and planar porphyrins. On the other hand,the NH chemical shift for compound 2 is more negative than 1 possibly due to the β-fluoro-substitution that increases the ring current effect. Variable-temperature 19F NMR spectroscopy for porphyrin 2 shows merge signals at 25 ℃,but two well-resolved porphyrin signals at low temperature (Figs. S3-S5 in Supporting information),indicating that faster inner NH proton exchange exist [27].
Single crystal of Zn-1 and Zn-2,suitable for X-ray diffraction analysis,were grown by the slow evaporation of a dichloromethane/ hexane solution. A perspective drawing from the crystal structure determination is shown in Fig. 1. The porphyrin core of Zn-1 and Zn-2 is perfectly planar. The bond lengths from the zinc ion to the pyrrole nitrogen atoms are in the range of 2.057(2)- 2.065(2)Å in Zn-2 and 2.038(4)-2.072(4)Å in Zn-1,comparable to that found in the literature [29, 30]. Moreover,the hexa-coordinated Zn atom is located in the mean plane of porphyrin ring in both Zn-1 and Zn-2 and is bound to two oxygen atoms from two carboxyl groups at the meso position of the porphyrin molecule. There is no solvent molecule in the crystal structure,and the plane of two adjacent porphyrins are parallel with a distance of 3.278Å for Zn-2 and 3.341Å for Zn-1 between the two planes. This indicates that there exits π···π stacking between the two adjacent porphyrin rings in Zn-1 and Zn-2. For Zn-2,the length of Zn-O bond is 2.272Å ,a little larger than the coordinate bond (2.07Å ),and is comparable to those found in literature for related species [30, 31, 32, 33]. Interestingly,this distance is somewhat smaller than the unfluorinated porphyrin Zn-1 (2.406Å ). The valence bond calculations show that the central zinc is present in its usual +2 oxidation state in both the porphyrins [34]. Each Zn-1 or Zn-2 unit links to two neighboring molecules through Zn-O bonds,which gives a condensed 1D chain coordination polymer assembly with a stepped pattern (Fig. S6 in Supporting information).
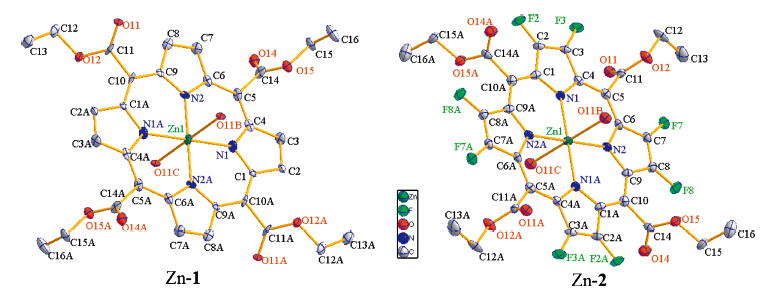
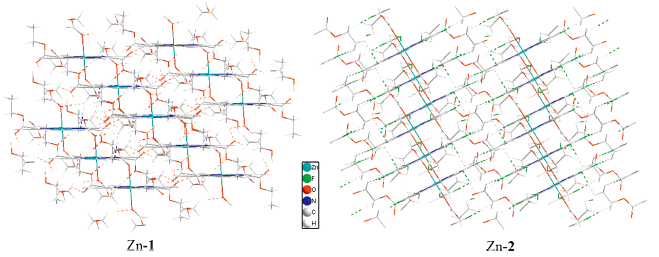
Hydrogen bonding plays an important role in the supramolecular assembly [35]. β-Octafluorination brings significant differences in the hydrogen bonding mode between Zn-1 and Zn-2. In Zn-1 crystal,there are two adjacent hydrogen bonds formed by side chain methyl hydrogen and ethoxy oxygen atom between two adjacent zinc porphyrin moieties within coordination polymer chain,and the 1D coordination polymer chains are assembled to 3D network by hydrogen bonding between methylene hydrogen atom of one polymer chain and coordinated carbonyl oxygen and nitrogen atom of other polymer chain (Fig. 2). The hydrogenbonding is,however,different in Zn-2 crystal. The two adjacent hydrogen bonds between the adjacent porphyrin moieties within the coordination polymer are formed by methyl hydrogen and bfluorine atom bonding. Furthermore,two opposite intramolecular hydrogen bonds formed by neighboring methyl hydrogen and fluorine atom exist in Zn-2. 3D network of Zn-2 complex is formed by the assembling of its 1D coordination polymer via hydrogen bonding of methylene hydrogen atom and β-fluorine atom between adjacent chains. The presence of β-fluorine atoms in Zn-2 gives more compactness to the whole crystal structure (Fig. 3). This is also evidenced by the fact that Zn-2 has significant higher density than Zn-1 (Table 1). Thus,β-octafluorination has important effect on the micro-environment of metal tetrakis( ethoxycarbonyl)porphyrin assembly.

|
Download:
|
| Fig 2. Hydrogen bonding (red dash line) of Zn-1 and Zn-2. (For interpretation of the references to color in this figure legend,the reader is referred to the web version of this article.) | |
{kind=link}

|
Download:
|
|
| Fig 3. 3D assembly packing drawing of Zn-1 and Zn-2. | ||
{kind=link}
In brief,we have synthesized meso-tetrakis(ethoxycarbonyl)- porphyrin and 2,3,7,8,12,13,17,18-octafluoro-meso-tetrakis( ethoxycarbonyl)porphyrin via acid-catalyzed condensation of ethyl glyoxylate with pyrrole and 3,4-difluoropyrrole,respectively. The free base porphyrins were transformed to their corresponding Zn(II) complexes whose well-resolved single crystal X-ray structures indicated that both the zinc porphyrins have distinct packing mode and hydrogen bonding. It was observed that the crystal structure of Zn-2 is more compact owing to the formation of hydrogen bonding between β-fluorine atom and hydrogen atom of alkyl side chains. An unusual six-coordination of Zn(II) was observed through intramolecular coordination of oxygen atoms of the two meso-substituents with central metal ion. β-Octafluorination caused blue shift in the absorption spectra and up-field shifted NH protons as well. Further studies on derivatization and applications of this novel meso-tetraalkyl-β-octafluoroporphyrin is underway.
AcknowledgmentThis work was supported by National Natural Science Foundation of China (NNSFC) under Grant (Nos. 21171057,21371059)
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.01.026.
| [1] | Y.S. Xie, K. Yamaguchi, M. Toganoh, et al., Triply N-confused hexaphyrins: nearinfrared luminescent dyes with a triangular shape, Angew. Chem. Int. Ed. 48 (2009) 5496-5499. |
| [2] | Y.S. Xie, P.C. Wei, X. Li, et al., Macrocycle contraction and expansion of a dihydrosapphyrin isomer, J. Am. Chem. Soc. 135 (2013) 19119-19122. |
| [3] | Y.Q. Wang, B. Chen, W.J. Wu, et al., Efficient solar cells sensitized by porphyrins with an extended conjugation framework and a carbazole donor: from molecular design to cosensitization, Angew. Chem. Int. Ed. 53 (2014) 10779-10783. |
| [4] | C.J. Li, Y.Q. Feng, X.J. Liu, T.Y. Zhang, The synthesis of porphyrin-anthraquinone dyad via an azo-rearrangement, Chin. Chem. Lett. 22 (2011) 539-542. |
| [5] | X.D. Li, Y.C. Zhu, L.J. Yang, Crown ether-appended Fe(III) porphyrin: synthesis, characterization and catalytic oxidation of cyclohexene with molecular oxygen, Chin. Chem. Lett. 23 (2012) 375-378. |
| [6] | J.E. Lyons, P.E. Ellis, H.K. Myers, Halogenated metalloporphyrin complexes as catalysts for selective reactions of acyclic alkanes with molecular oxygen, J. Catal. 155 (1995) 59-73. |
| [7] | D. Dolphin, T.G. Traylor, L.Y. Xie, Polyhaloporphyrins: unusual ligands for metals and metal-catalyzed oxidations, Acc. Chem. Res. 30 (1997) 251-259. |
| [8] | W.J. Su, T.M. Cooper, M.C. Brant, Investigation of reverse-saturable absorption in brominated porphyrins, Chem. Mater. 10 (1998) 1212-1213. |
| [9] | R. Bonnett, I.A.D. Gale, G.F. Stephenson, The meso-reactivity of porphyrins and related compounds. Part II. Halogenation, J. Chem. Soc. (C) Org. (1966) 1600-1604. |
| [10] | D. Mandon, P. Ochenbein, J. Fischer, et al., β-Halogenated-pyrrole porphyrins. Molecular structures of 2,3,7,8,12,13,17,18-octabromo-5,10,15,2'-tetramesitylporphyrin, nickel(II) 2,3,7,8,12,13,17,18-octabromo-5,10,15,2'-tetramesitylporphyrin, and nickel(II) 2,3,7,8,12,13,17,18-octabromo-5,10,15,2'-tetrakis(pentafluorophenyl) porphyrin, Inorg. Chem. 31 (1992) 2044-2049. |
| [11] | K.A. Nguyen, P.N. Day, R. Pachter, Effects of halogenation on the ionized and excited states of free-base and zinc porphyrins, J. Chem. Phys. 110 (1999) 9135- 9144. |
| [12] | S. Evans, J.R.L. Smith, The oxidation of ethylbenzene and other alkylaromatics by dioxygen catalysed by iron(III) tetrakis(pentafluorophenyl)porphyrin and related iron porphyrins, J. Chem. Soc., Perkin Trans. 2 (2000) 1541-1552. |
| [13] | K.A. Nguyen, P.N. Day, R. Pachter, et al., Analysis of absorption spectra of zinc porphyrin, zinc meso-tetraphenylporphyrin, and halogenated derivatives, J. Phys. Chem. A 106 (2002) 10285-10293. |
| [14] | J. pyrimidines and pyrimido2015-5-25 β-Fluorinated porphyrins and related compounds: an overview, Eur. J. Org. Chem. 2008 (2008) 417-433. |
| [15] | H.Y. Liu, T.S. Lai, L.L. Yeung, C.K. Chang, First synthesis of perfluorinated corrole and its MnO complex, Org. Lett. 5 (2003) 617-620. |
| [16] | E. Steene, A. Dey, A. Ghosh, β-Octafluorocorroles, J. Am. Chem. Soc. 125 (2003) 16300-16309. |
| [17] | J. Leroy, C. Wakselman, First access to 3,4-difluoro-1H-pyrrole, Tetrahedron Lett. 35 (1994) 8605-8608. |
| [18] | A.J.F.N. Sobral, A.M.D.A.R. Gonsalves, The manganese complex of 2,3,7,8,12,13,17,18-octaphenylporphyrin as epoxidation catalyst, J. Porphyrins Phthalocyan. 5 (2001) 428-430. |
| [19] | A.J.F.N. Sobral, A.M.D.A.R. Gonsalves, 5,15-Diaryl-β-substituted-porphyrinatomanganese( III) chlorides as probes for structure-activity relationships in porphyrin- based epoxidation catalysts, J. Porphyrins Phthalocyan. 5 (2001) 861-866. |
| [20] | C. Paliteiro, A. Sobral, Electrochemical and spectroelectrochemical characterization of meso-tetra-alkyl porphyrins, Electrochim. Acta 50 (2005) 2445-2451. |
| [21] | S. Neya, J.S. Quan, M. Hata, T. Hoshino, N. Funasaki, A novel and efficient synthesis of porphine, Tetrahedron Lett. 47 (2006) 8731-8732. |
| [22] | M.O. Senge, M. Davis, Porphyrin (porphine)—a neglected parent compound with potential, J. Porphyrins Phthalocyan. 14 (2010) 557-567. |
| [23] | M.P. Trova, P.J.F. Gauuan, A.D. Pechulis, et al., Superoxide dismutase mimetics. Part 2: Synthesis and structure-activity relationship of glyoxylate- and glyoxamide- derived metalloporphyrins, Bioorg. Med. Chem. 11 (2003) 2695-2707. |
| [24] | X.L. Yang, C.D. Wu, Metalloporphyrinic framework containing multiple pores for highly efficient and selective epoxidation, Inorg. Chem. 53 (2014) 4797-4799. |
| [25] | J.S. Lindsey, I.C. Schreiman, H.C. Hsu, P.C. Kearney, A.M. Marguerettaz, Rothemund and Adler-Longo reactions revisited: synthesis of tetraphenylporphyrins under equilibrium conditions, J. Org. Chem. 52 (1987) 827-836. |
| [26] | M. Gouterman, Spectra of porphyrins, J. Mol. Spectrosc. 6 (1961) 138-163. |
| [27] | E.K. Woller, S.G. DiMagno, 2,3,7,8,12,13,17,18-Octafluoro-5,10,15,2'-tetraarylporphyrins and their zinc complexes: first spectroscopic, electrochemical, and structural characterization of a perfluorinated tetraarylmetalloporphyrin, J. Org. Chem. 62 (1997) 1588-1593. |
| [28] | J. Leroy, A. Bondon, L. Toupet, C. Rolando, 2,3,7,8,12,13,17,18-octafluoro- 5,10,15,2'-tetraphenylporphyrin: first synthesis and X-ray crystal structure of the ZnII complex, Chem. Eur. J. 3 (1997) 1890-1893. |
| [29] | V.V. Smirnov, E.K. Woller, D. Tatman, S.G. DiMagno, Structure and photophysics of β-octafluoro-meso-tetraarylporphyrins, Inorg. Chem. 40 (2001) 2614-2619. |
| [30] | W. Chen, S. Fukuzumi, Change in supramolecular networks through in situ esterification of porphyrins, Eur. J. Inorg. Chem. 2009 (2009) 5494-5505. |
| [31] | M.P. Byrn, C.J. Curtis, Y. Hsiou, et al., Porphyrin sponges: conservative of host structure in over 200 porphyrin-based lattice clathrates, J. Am. Chem. Soc. 115 (1993) 9480-9497. |
| [32] | P. Bhyrappa, S.R. Wilson, K.S. Suslick, Hydrogen-bonded porphyrinic solids: supramolecular networks of octahydroxy porphyrins, J. Am. Chem. Soc. 119 (1997) 8492-8502. |
| [33] | R.K. Kumar, S. Balasubramanian, I. Goldberg, Supramolecular multiporphyrin architecture. coordination polymers and open networks in crystals of tetrakis( 4-cyanophenyl)- and tetrakis(4-nitrophenyl)metalloporphyrin, Inorg. Chem. 37 (1998) 541-552. |
| [34] | M. O’Keefe, N.E. Brese, Atom sizes and bond lengths in molecules and crystals, J. Am. Chem. Soc. 113 (1991) 3226-3229. |
| [35] | Q.Y. Liu, Q.Y. Jia, J.Q. Zhu, et al., Highly ordered arrangement of meso-tetrakis(4- aminophenyl)porphyrin in self-assembled nanoaggregates via hydrogen bonding, Chin. Chem. Lett. 25 (2014) 752-756. |