




Jin Yin Hua,the flower buds of Lonicera japonica Thunb. (caprifoliaceae),is a common ingredient of formulations used in traditional Chinese medicine for treating influenza,cold,fever,and infections [1]. Chemical and pharmacological studies resulted in characterization of constituents with different structural features and biological activities [2, 3, 4, 5, 6, 7, 8]. As part of a programme to assess the chemical and biological diversity of traditional Chinese medicines [9, 10, 11],we conducted detailed chemical analysis of an aqueous extract of the flower buds of L. japonica,since the flower bud decoction is practically used. Our previous studies on the aqueous extract led to the isolation of homosecoiridoids having structural characters of the secoiridoid nucleus coupled with N-substituted nicotinic acid or pyridine units (lonijaposides A-W),phenylpyruvic acid derived moieties (loniphenyruviridosides A-D),and β-hydroxy amino acid units [12, 13, 14, 15],as well as two N-(6-Oacyl- β-D-glucopyranosyl)-nicotinate (lonijaponinerosides A and B) [16]. In addition,after the flower buds were extracted by water,the residue was further extracted with EtOH (95%),from which six new aromatic glycosides and 48 known compounds were characterized [17, 18]. Some of these compounds showed antiviral activity against the influenza virus A/Hanfang/359/95 (H3N2) and Coxsackie virus B3 replication,as well as antiinflammatory activity against the release of glucuronidase in rat polymorphonuclear leukocytes induced by the platelet-activating factor and STAT-3 (signal transducers and activators of transcription 3) inhibitory activity. Continuing investigation on the aqueous extract,two new homosecoiridoids loniaceticiridoside (1) and lonimalondialiridoside (2) have been isolated. We report herein the isolation,structure elucidation,and biological activity of the two compounds. 2. Experimental 2.1. General experimental procedures
Optical rotations were measured on a PE Model 343. UV spectra were measured on a JASCO J-810 spectropolarimeter. IR spectra were recorded on a Nicolet 5700 FT-IR Microscope spectrometer (FT-IR Microscope Transmission). 1D- and 2D-NMR spectra were obtained at 500 MHz or 600 for 1H and 125 or 150 MHz for 13C, respectively,on an Inova 500 MHz or Bruker AV III spectrometer with solvent peaks as references (unless otherwise noted). ESIMS data were measured with a Q-Trap LC/MS/MS (Turbo Ionspray source) spectrometer. HR-ESIMS data were,in turn,measured on an AccuToFCS JMS-T100CS spectrometer. Column chromatography was performed with silica gel (200-300 mesh,Qingdao Marine Chemical Inc.,Qingdao,China) and Pharmadex LH-20 (Pharmacia Biotech AB,Uppsala,Sweden). HPLC separation was performed on an instrument with a Waters 600 controller,a Waters 600 pump, and a Waters 2487 dual λ absorbance detector on an Prevail (250 mm×10 mmi.d.) semi-preparative column packed with C18 (5 μm). Glass precoated silica gel GF254 plates were used for TLC. Spots were visualized under UV light or by spraying with 7% H2SO4 in 95% EtOH followed by heating. 2.2. Plant material See Ref. [12]. 2.3. Extraction and isolation
For extraction and preliminary fractionation of the extract,see Refs. [14, 15]. Fraction B4 (86 g) was chromatographed over a RP silica gel column,eluting with a gradient of EtOH (0-100%) in H2O, to yield subfractions B4-1-B4-7,of which subfraction B4-4 (8.5 g) was further separated by flash chromatography over RP silica gel, eluting with a gradient of MeOH (0-50%) in H2O to give subfractions (B4-4-1-B4-4-10). B4-4-4 (290 mg) was subjected to RP-HPLC,using CH3OH-H2O (35:65) as the mobile phase,to afford 2 (2.5 mg,0.000021%),and B4-4-9 (61 mg) was separated by RP-HPLC,using CH3CN-H2O (12:88) containing 0.5% HOAc as the mobile phase,to yield 1 (6.6 mg,0.000055%).
Loniaceticiridoside (1): White amorphous powder,soluble in H2O,MeOH,and EtOH; [α]D 20 -203.2 (c 0.38,H2O); UV (H2O) λmax (log ε) 246 (3.94) nm; CD (H2O) 200 (△ε -6.14),245 (△ε -6.75) nm; IR νmax 3383,2923,1691,1614,1390,1358, 1320,1279,1205,1063,918,837,759,637,609 cm-1; 1H NMR (D2O,500 MHz),see Table 1; 13C NMR (D2O,125 MHz),see Table 1; (+)-ESIMS m/z 417 [M+H]+,439 [M+Na]+,455 [M+K]+; HR-ESIMS m/z 417.1390 [M+H]+ (calcd. for C18H25O11 417.1397).
| Table 1 NMR spectroscopic data for compounds 1 and 2a. |
Lonimalondialiridoside (2): White amorphous powder,soluble inH2O,MeOH,and EtOH; [α]D20-95.0 (c 0.12,H2O); UV (H2O) λmax (log ε) 241 (3.73) nm; CD (H2O) 225.5 (△ε -51.67),257.5 (△ε +23.11) nm; IR νmax 3372,2937,1728,1697,1608,1412,1374, 1288,1231,1185,1042,982,914,890,860,820,782,767, 735 cm-1; 1H NMR (D2O,600 MHz),see Table 1; 13C NMR (D2O, 150 MHz),see Table 1; (+)-ESIMS m/z 465 [M+Na]+,481 [M+K]+;HR-ESIMS m/z 465.1388 [M+Na]+ (calcd. for C20H26O11Na 465.1373). 2.4. Enzymatic hydrolysis of 1
An aqueous solution (1 mL) of compound 1 (4 mg) was treated with β-glucosidase from almonds (15 mg,8.92 U/mg,Mw135,000, Sigma-Aldrich Corporation,USA) at 37 ℃ for 12 h. The reaction mixture was extracted with EtOAc (2 mL×2). The H2O phase was concentrated to dryness under reduced pressure,and the residue was chromatographed over silica gel,eluting with CH3CN-H2O (6:1),to yield glucose with [α]D20 40.5 (c 0.09,H2O). The solvent system CH3CN-H2O (4:1) was used for TLC identification of glucose (Rf = 0.39). 2.5. Assays for pharmacological activities of 1 and 2
Details may be found in Refs. [12, 13, 14] and the references cited therein. 3. Results and discussion
Compound 1 was obtained as a white amorphous solid,[α]D20 -203.2 (c 0.38,H2O). Its IR spectrum showed absorption bands for hydroxy (3383 cm-1) and conjugated carbonyl (1691 and 1613 cm-1) functionalities. The positive ESIMSof 1 exhibited pseudo molecular ion peaks at m/z 417 [M+H]+,439 [M+Na]+,and 455 [M+K]+. HR-ESIMS at m/z 417.1390 [M+H]+ indicated the molecular formula as C18H24O11 (calcd. for C18H25O11,417.1397), which was supported by the NMR spectroscopic data (Table 1). The 1H NMRspectrum of 1 in CD3OD showed resonances attributed to a deshielded trisubstituted olefinic proton at δH 7.54 (d,J = 2.5 Hz, H-3),an acetalic proton at δH 5.50 (d,J = 1.5 Hz,H-1),a vinyl group at δH 5.47 (ddd,J = 17.5,10.5,and 9.5 Hz,H-8),5.25 (dd,J = 17.5 and 1.5 Hz,H-10a),and 5.21 (d,J = 10.5 and 1.5 Hz,H-10b),and an oxymethine δH 4.76 (m,H-7). It also showed resonances due to two methines at δH 3.15 (m,H-5) and 2.64 (ddd,J = 9.5,5.5,and 1.5 Hz, H-9),and a methylene at δH 1.88 (ddd,J = 13.5,3.5,and 3.5 Hz, H-6a) and 1.41 (ddd,J = 13.5,13.5,and 11.5 Hz,H-6b). In addition, the 1H NMR spectrumof 1 displayed characteristic resonances due to a β-glucopyranosyl unit (Table 1) and a methylene unit attached to a methine at δH 2.61 (dd,J = 15.5 and 7.0 Hz,H-200a) and 2.53 (dd, 1H,J = 15.5 and 5.5 Hz,H-200b). The presence of β-glucopyranosyl unit was confirmed by enzymatic hydrolysisof 1 with bglucosidase, which produced glucose identified on the basis of TLC by comparing with an authentic sugar sample. The glucose isolated from the hydrolysate gave a positive optical rotation, [α]D20 40.5 (c 0.09,H2O),indicating that it was the D-glucose [12, 13]. Besides carbon resonances corresponding to the above units,the 13C NMR and DEPT spectra of 1 showed two carbonyl resonances (Table 1) at δC 174.9 (broad and low intensity) and 168.3,respectively. These NMR spectroscopic data were very similar to those of the co-occurring secologanic acid [13] except that the acetalic C-7 in secologanic acid was replaced by the oxymethine in 1 and that there were additional methylene and carboxylic units in 1. This combined with the HR-ESIMS data suggested that 1 was an abnormal homosecoiridoid β-D-glucoside, which was confirmed by analysis of the 2D NMR data.

|
Download:
|
| Fig 1. Structures of compounds 1 and 2. | |
{kind=link}
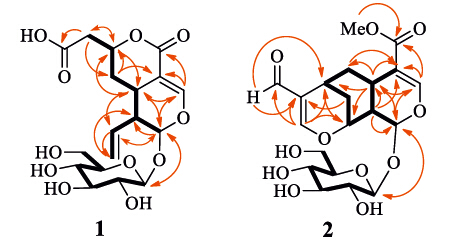
The proton and protonated carbon signals in the NMR spectra of 1 were assigned by the gHSQC experiment. In the 1H-1H COSY spectrum of 1,homonuclear vicinal coupling correlations H- 1↔H-9↔H-5↔H2-6↔H-7↔H2-2′′ and H-9↔H-8↔H2-10, together with the chemical shifts and coupling constants of these protons,revealed the presence of the secoiridoid parent structure moiety with a 8(10)-olefonic functional unit and an extension of the methylene unit at C-7 (Fig. 2,thick lines). This was verified by two- and three-bond heteronuclear correlations (Fig. 2,red arrows) in the HMBC spectrumof 1: H-1/C-3,C-5,C-8,and C-9; H-3/C-1,C-4,C-5,and C-11; H-5/C-1 and C-7; H2-6/C-4,C-5,C-7, and C-9; H-7/C-2′′ and C-5; H-8/C-1,C-5,C-9,and C-10; H-9/C-1, C-4,C-5,C-6,C-8,and C-10; H2-10/C-8 and C-9; and H2-2′′/C-6 and C-7. In addition,gCOSY cross-peaks H-1′ ↔H-2′ ↔H-3′ ↔H- 4′ ↔H-5′ ↔H2-6′ and HMBC correlations H-1/C-10 and H-10/C-1 confirmed the presence of the β-D-glucopyranosyl unit at C-1 in 1. The HMBC correlations H-2′′/C-1′′ ,together with their shift,located the carboxylic group at C-2′′ ,which was supported by the distinctively broadened C-1′′ and C-2′′ resonances possibly caused by a dynamic conjugation of the free carboxylic acid group in 1. Because the H-7 resonance intensity was significantly diminished by coupling with H2-2′′ and H2-6,the HMBC correlation from H-7 to C-11 was not observed. However,the shifts of H-3 and H-7 and C-3,C-4,and C-7 [12, 13, 14],together with molecular formula, demonstrated a lactone formation between the oxymethine unit (c-7) and the carbonyl group (c-11) in 1. Therefore,the planar structure of compound 1 was elucidated as shown in Fig. 2.

|
Download:
|
| Fig 2. Main 1H–1H COSY (black thick lines) and HMBC correlations (red arrows, from 1H to 13C) of 1 and 2. | |
{kind=link}
In the NOE difference spectrum (Figs. S13 and S14 in Supporting information),irradiation of H-5 enhanced H-6a,H-7,and H-9; irradiation of H-9 caused enhancements of H-5 and H-6a; and in turn irradiation of H-6a gave enhancements of H-5,H-7,and H-9. These enhancements revealed that H-5,H-6a,H-7,and H-9 were cofacial. In addition,the enhancements of H-8 and H2-2′′ upon irradiation of H-6b indicated that these protons were cofacial on other side of the ring system. Similarity of the CD spectra between 1 and the co-occurring secologanic acid and sweroside (Figs. S15-S17 in Supporting information) suggests that the secoiridoid nucleus of the these analogues possesses the same absolute configuration,of which the configuration of secologanic acid was determined by a single-crystal X-ray crystallographic analysis using anomalous scattering of Cu Kα radiation [13]. Therefore,the structureof 1 was determined and designated as loniaceticiridoside.
Compound 2,a white amorphous solid with [α]D20 -95.0 (c 0.12,H2O),showed IR absorption bands for hydroxy (3372 cm-1) and conjugated carbonyl (1728,1697,and 1608 cm-1) functional groups. The molecular formula C20H26O11 was indicated by HR-ESIMS at m/z 465.1388 [M+Na]+ (calcd. for C20H26O11Na 465.1373) and the NMR spectroscopic data (Table 1). Comparison of the NMR spectroscopic data of 2 and 1 indicated replacement of the C-1′′ and C-2′′ unit in 1 by a 2′′-substituted 3′′-oxypropenal moiety in 2 {δH 8.99 (s,1H,H-1′′) and 7.54 (s,1H,H-3′′); δC 195.7 (c-1′′),125.2 (c-2′′),and 174.5 (c-3′′)},and the presence of a methoxy group (δH 3.68 and δC 54.7). In addition,the vinyl group in 1 was substituted by sp3 hybrid methine and methylene units {δH 2.81 (m,1H,H-7),2.00 (brd,1H,J = 14.4 Hz,H-10a),and 1.86 (m,1H,H-10b); δC 23.9 (c-7) and 31.3 (c-10)}. This suggested that compound 2 was another uncommon homoiridoid β-glucopyranoside, which was further elucidated by 2D NMR data analysis. The 1H-1H gCOSY spectrum showed cross peaks of H-1↔ H-9↔H-5↔H2-6↔H-7↔H2-10↔H-8↔H-9 and H-1′ ↔ H-2′ ↔H-3′ ↔H-4′ ↔H-5′ ↔H2-6′ (Fig. 2,thick lines),indicating the presence of vicinal coupling chain extensions in 2. In the gHMBC spectrum,correlations from H-1 to C-1′ ,C-3 and C-5; from H-3 to C-1,C-4,C-5,and C-11; from H-9 to C-1,C-4,and C-5; and from OCH3 to C-11; together with chemical shifts of these proton and carbon resonances and a W-type coupling 1H-1H gCOSY correlation H-3↔H-5,confirmed the presence of the same 1-bglucopyranosyl cyclohexenolic ether moiety in 2 as that in 1. The gHMBC correlations from H-1 to C-8; from H-5 to C-4,C-6,C-7,C-8, and C-9; and from H-6 to C-4; in combination with an extension of vicinal coupling correlations H-1↔H-9↔H-5↔H2-6↔H- 7↔H2-10↔H-8↔H-9 in the 1H-1H gCOSY spectrum,demonstrated that the cyclohexenolic ether moiety was fused through C-5 and C-9 with a 7,8-disubstituted six-membered ring. In addition,the gHMBC correlations from H-10 to C-2′′; from H-1′′ to C-200 and C-7; and from H-3′′ to C-1′′,C-2′′ ,C-7,and C-8 revealed that the 3′′-oxypropenal moiety was connected through C-2′′ and 300-oxy to C-7 and C-8,respectively. Therefore,the planar structure of compound 2 is elucidated as shown,which is an unusual homoiridoid β-glucopyranoside with an additional cyclohexenolic ether moiety.
The configuration of 2 was deduced from comprehensive analysis of the NOE difference experiment,the coupling constant values,and the CD spectrum. In the NOE difference spectrum of 2, irradiation of H-1 enhanced H-8 and H-9,and the H-6b and H-9 resonances were enhanced by irradiation of H-5. In addition,when H-7 was irradiated,H-6b and H-10b were enhanced,and the H-9 and H-10a resonances were enhanced upon irradiation of H-8. These enhancements,together with the coupling constants between H-5 with H2-6 and H-9 (J5,6a ≈ 0 Hz,J5,6b = 6.6 Hz,and J5,9 = 7.8 Hz),between H-7 (brs,W1/2 ≈ 7.8 Hz) with H2-6 and H2- 10,and between H-8 (brs,W1/2 ≈ 7.8 Hz) with H-9 and H2-10, demonstrated that the cyclohexane ring in 2 had a chair conformation with H-6b and H-9 at axial and H-5,H-6a,H-7, and H-8 at equatorial positions (Fig. 3). The CD spectrum of 2 displayed typical coupled Cotton effects,positive at 257.5 nm and negative at 225.5 nm (corresponding to an UV absorption at 241 nm),arising from the interaction between π→π* transition moments of the two carbonyl-conjugated cyclohexenolic chromophores in the molecule. This was confirmed by comparison of the CD spectrum of 2 with those of the co-occurring secoiridoids, e.g. secologanic acid and sweroside. Based on the CD exciton chirality method [19, 20],the absolute configuration at C-7 and C-8 of 2 was assigned as 7R,8R. This,combined with the aforementioned NOE enhancements and a biogenetic postulation,the absolute configuration of 2 were assigned as shown in Fig. 1. Therefore,the structure of compound 2 was determined and designated as lonimalondialiridoside.

|
Download:
|
| Fig 3. Main NOE enhancements (blue double arrows) of 1 and 2. | |
{kind=link}
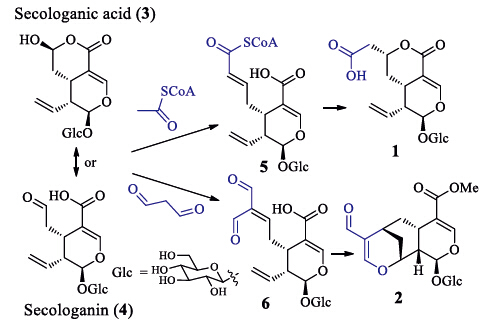
The biosynthetic precursorsof 1 and 2 are proposed to be the co-occurring secologanic acid (3,abundant in L. japonica) and/or secologanin (4) [12, 13]. A plausible pathway to compounds 1 and 2 are postulated in Scheme 1. Condensation of the precursor with the basic biosynthetic unit acetyl-CoA or malondialdehyde by an aldollike reaction would yield key intermediates (5 or 6) with a carbonyl-conjugated double bond. The intermediate 5 undergoes 1,2-addition of the conjugated double with the carboxylic alcohol to generate 1,while the intermediate 6 may be cyclized through an intramolecular hetero-Diels-Alder addition to yield 2. As the absolute configurations of secologanic acid was determined by a single-crystal X-ray crystallographic analysis using anomalous scattering of Cu Ka radiation [13],the biosynthetic pathways support the assignment of the absolute configurations for 1 and 2. Adinoside A from Adina racemosa [21] and stryspinoside from Strychnos spinosa [22] are the only known natural products possessing the carbon skeletonsof 1 and 2,reported from species rather than L. japonica. Both the known compounds were also isolated from the ethanol extract of the L. japonica flower buds [18], and an ester of adinoside A coupling with a phenolic glucoside (lonicerjaponin A) from a methanol extract [24]. On the basis of structural features,compounds 1 and 2 are proposed to be biosynthetic intermediates of adinoside A and lonicerjaponin A and stryspinoside,respectively. In addition,a compound with the same skeleton as that of 2 and stryspinoside were synthesized from secologanin through a tandem-Knoevenagel-hetero-Diels-Alder reaction [23]. This supports the postulated biogenetic pathway for the two types of homosecoiridoids.

|
Download:
|
| Scheme 1. The plausible biosynthetic pathway of 1 and 2. | |
{kind=link}
In preliminary in vitro assays,compounds 1 and 2,at10 μmol/L, showed inhibitory activity against the release of glucuronidase in rat polymorphonuclear leukocytes induced by the plateletactivating factor,with inhibition rates of (66.4±5.1)% and (72.6±3.8)%,respectively,while the positive control (ginkgolide B) exhibited an inhibition rate of (71.0±4.5)% at the same concentration [12]. The compounds were also assessed for their activities against influenza virus A/Hanfang/359/95 (H3N2),Coxsackie virus B3, and HIV-1 replication,as well as against several human cancer cell lines,but all were inactive at a concentration of 10 μmol/L. 4. Conclusion
Two new homosecoiridoids with anti-inflammatory activity, named loniaceticiridoside (1) and lonimalondialiridoside (2),were isolated from the aqueous extract of L. japonica flower buds. Their unique structures were elucidated by comprehensive spectroscopic data analysis. Compounds 1 and 2 are second examples of natural products with corresponding skeletons. In particular,the plausible biosynthetic pathway associated to the different types of co-occurring iridoids [12, 13, 14, 15, 16, 17, 18, 23] provides an important clue for further studies of biomimetic and total synthesis,chemical transformation,structural modification,and structure-activity relationships,as well as biosynthesis of the diverse iridoids from the flower buds of L. japonica.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (NNSFC; Nos. 20772156 and 30825044),the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT,No. IRT1007),and the National Science and Technology Project of China (Nos. 2012ZX09301002-002 and 2011ZX0 9307- 002-01) is acknowledged.
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2014.11.035.
| [1] | Jiangsu New Medical College, Dictionary of Traditional Chinese Medicine, Shanghai Science and Technology Publishing House, Shanghai, 1977, pp. 1403-1405. |
| [2] | R.W. Teng, D.Z. Wang, C.X. Chen, Two triterpenoid saponins from Lonicera japonica, Chin. Chem. Lett. 11 (2000) 337-340. |
| [3] | R. Kakuda, M. Imai, Y. Yaoita, M. Koichi, K. Masao, Secoiridoid glycosides from the flower buds of Lonicera japonica, Phytochemistry 55 (2000) 879-881. |
| [4] | C.W. Choi, H.A. Jung, S.S. Kang, J.S. Choi, Antioxidant constituents and a new triterpenoid glycoside from Flos Lonicerae, Arch. Pharm. Res. 30 (2007) 1-7. |
| [5] | D.Q. Yu, R.Y. Chen, L.J. Huang, et al., The structure and absolute configuration of Shuangkangsu: a novel natural cyclic peroxide from Lonicera japonica (Thunb.), J. Asian Nat. Prod. Res. 10 (2008) 851-856. |
| [6] | L.M. Lin, X.G. Zhang, J.J. Zhu, et al., Two new triterpenoid saponins from the flowers and buds of Lonicera japonica, J. Asian Nat. Prod. Res. 10 (2008) 925-929. |
| [7] | E.J. Lee, J.S. Kim, H.P. Kim, J.H. Lee, S.S. Kang, Phenolic constituents from the flower buds of Lonicera japonica and their 5-lipoxygenase inhibitory activities, Food Chem. 120 (2010) 134-139. |
| [8] | Z.F. Zheng, Q.J. Zhang, R.Y. Chen, D.Q. Yu, Four new N-contained iridoid glycosides from flower buds of Lonicera japonica, J. Asian Nat. Prod. Res. 14 (2012) 729-737. |
| [9] | M.H. Chen, L. Lin, L. Li, et al., Enantiomers of an indole alkaloid containing unusual dihydrothiopyran and 1,2,4-thiadiazole rings from the root of Isatis indigotica, Org. Lett. 14 (2012) 5668-5671. |
| [10] | Y. Tian, Q.L. Guo, W.D. Xu, et al., A minor diterpenoid with a new 6/5/7/3 fusedring skeleton from Euphorbia micractina, Org. Lett. 16 (2014) 3950-3953. |
| [11] | W.D. Xu, Y. Tian, Q.L. Guo, Y.C. Yang, J.G. Shi, Secoeuphoractin, a minor diterpenoid with a new skeleton from Euphorbia micractina, Chin. Chem. Lett. 25 (2014) 1531-1534. |
| [12] | W.X. Song, S. Li, S.J. Wang, et al., Pyridinium alkaloid-coupled secoiridoids from the flower buds of Lonicera japonica, J. Nat. Prod. 71 (2008) 922-925. |
| [13] | Y. Yu, W.X. Song, C.G. Zhu, et al., Homosecoiridoids from the flower buds of Lonicera japonica, J. Nat. Prod. 74 (2011) 2151-2160. |
| [14] | Y. Yu, C.G. Zhu, S.J. Wang, et al., Homosecoiridoid alkaloids with amino acid units from the flower buds of Lonicera japonica, J. Nat. Prod. 76 (2013) 2226-2233. |
| [15] | W.X. Song, Y.C. Yang, J.G. Shi, Two new β-hydroxy amino acid-coupled secoiridoids from the flower buds of Lonicera japonica: isolation, structure elucidation, semisynthesis, and biological activities, Chin. Chem. Lett. 25 (2014) 1215-1219. |
| [16] | Z.B. Jiang, W.X. Song, J.G. Shi, Two new 1-(6'-O-acyl-β-D-glucopyranosyl)pyridinium- 3-carboxylates from the flower buds of Lonicera japonica, Chin. Chem. Lett. 26 (2015) 69-72. |
| [17] | F. Wang, Y.P. Jiang, X.L. Wang, et al., Aromatic glycosides from the flower buds of Lonicera japonica, J. Asian Nat. Prod. Res. 15 (2013) 492-501. |
| [18] | F. Wang, Y.P. Jiang, X.L. Wang, et al., Chemical constituents from flower buds of Lonicera japonica, China J. Chin. Mater. Med. 38 (2013) 1378-1385. |
| [19] | N. Harada, K. Nakanishi, Determining the chiralities of optically active glycols, J. Am. Chem. Soc. 91 (1969) 3989-3991. |
| [20] | M. Koreeda, N. Harada, K. Nakanishi, Exciton chirality methods as applied to conjugated enones, esters, and lactones, J. Am. Chem. Soc. 96 (1974) 266-268. |
| [21] | A. Itoh, K. Fujii, S. Tomatsu, et al., Six secoiridoid glucosides from Adina racemosa, J. Nat. Prod. 66 (2003) 1212-1216. |
| [22] | A. Itoh, N. Oya, E. Kawaguchi, et al., Secoiridoid glucosides from Strychnos spinosa, J. Nat. Prod. 68 (2005) 1434-1436. |
| [23] | L.F. Tietze, C. Bärtels, Synthesis of bridged homoiridoids from secologanin by tandem-Knoevenagel-hetero-Diels-Alder reactions, Liebigs Ann. Chem. 1991 (1991) 155-160. |
| [24] | Y. Kashiwada, Y. Omichi, S. Kurimoto, et al., Conjugates of a secoiridoid glucoside with a phenolic glucoside from the flower buds of Lonicera japonica Thunb, Phytochemistry 96 (2013) 423-429. |