




b Department of Biophysics, University of Michigan, Ann Arbor, MI 48109, USA
The behavior and stability of proteins at solid interfaces are keyfactors influencing the performance of many technologies such asprotein-based biosensors,protein microarrays,and medicalimplants [1, 2, 3, 4, 5, 6, 7]. Many protein-based biosensors have been developed because of their potential advantages including high costefficiency,good portability,and rapid-response potency. However,the optimal performance of a number of current biosensors is notfully achieved for practical use due to the lack of predictablebehavior of proteins on the chip surface [8]. The activity of aprotein on a chip is highly dependent upon its stability changeinduced by the surface potential and its orientation affected bysurface chemistry. Based on knowledge of such surface-proteininteractions,several efforts have been reported to achieveimproved performance of protein-based biosensors,includingsolid surface coating [9, 10],specific tethering [11, 12],and solventadjustment [13]. Despite the success in such examples,fundamental knowledge of protein-surface interactions at residue-levelresolution is still lacking,so that reasonable design of proteinbased biosensors is far from routine application.
As shown by previous research on protein-surface interactions,itis difficult to capture high resolution structural details of proteinsand peptides on surfaces using experimental methods such ascircular dichroism (CD),attenuated total reflection Fourier transform IR (ATR-FTIR),2-dimentional nuclear magnetic resonance(NMR),and atomic force microscopy (AFM) [4, 14, 15, 16, 17, 18, 19],due to eitherlow resolution or limit in ability for applications to surface attachedproteins [20]. Therefore,alternative methods are needed to capturethe behavior of proteins on surfaces in residue-level detail.Molecular simulation may be able to bridge this gap due to itsinherent competence in investigating detailed structures. Suchapproaches have been successfully applied to studies of protein-surface interactions [21, 22]. Atomistic simulations provide atomiclevel structural details of the peptide,but it is difficult to measure thestability of peptides due to large sampling requirements in bothfolded and unfolded structures. Therefore,coarse grained simulationmethods are more favorable to meet the sampling needs by reducingthe number of degrees of freedom. Many studies have shown successin understanding protein orientation on charged or non-chargedself-assembled monolayer surfaces with different hydrophobicity[23, 24, 25, 26, 27, 28, 29]. Coarse grained force field was further developed forprotein-membrane interactions and solvent conditions [30, 31]However,to capture the mechanism of protein-surface interactionsrequires a sophisticated protein model that can reproduce theprotein folding mechanism and a well parameterized and validatedsurface potential with which to couple it. Such a coarse grainedmodel has been previously built based on the Karanicolas andBrooks’ Go-like protein model and extensions implementing a wellparameterized surface force field [32]. This model can capture thestability and structure of a protein when it is either physicallyadsorbed on or chemically tethered to a surface with a specifichydrophobic character.
By using such a coarse grained simulation method,we wouldlike to understand the detailed structures of a peptide thatinteracts with a surface and how the simulation can guide thedesign of the peptide for a desired behavior on the surface. Toachieve this goal,a simple peptide-surface model system isstudied. Specifically,cecropin P1,a small antimicrobial peptide,isused. This peptide has been widely used both experimentally andcomputationally as a model peptide for biosensor design due to itssmall size,simple secondary structure,and clear application foractivity detection [13]. The sequence of this peptide is composed of32 residues with various hydrophobicities. Basically,hydrophobicresidues are dilutely scattered along the sequence,which providepotential sites for hydrophobic interactions with membraneswhen a helix structure formed. There is one region near the Cterminus formed by 5 continuous hydrophobic residues (residue22-26),which is identified as the part that penetrates the bacterialcell membrane. The formation of secondary structure in cecropinP1 is dependent on the solvent and surface conditions. In purewater or phosphate buffer solution (PBS),cecropin P1 has arandom-coil structure,but when interacting with membranes or inhydrophobic solution [33, 34] it forms an a-helix. Previousexperimental work using sum frequency generation (SFG)vibrational spectroscopy by Chenet al. [13, 35, 36] has shown theinteresting results that cecropin P1 forms different secondarystructures or employs different surface orientations under varioussolvent conditions and on maleimide terminated self-assembledmonolayer (SAM) surfaces.
This work examines the interaction between cecropin P1 and amaleimide self-assembled monolayer surface,which is consistentwith the experimental set-up of Hanet al. [13] In their work,twomain findings were: (a) the peptide forms a better helical structureon the surface than in the bulk solution; (b) the peptide hasdifferent orientations on the maleimide SAM surface by tetheringthe peptide with different terminal residues. By using coarsegrained simulation methods,in this work,we explore the thermalstability of cecropin P1 in the bulk and on surfaces. The surfacetethered peptides show largely improved stability compared withpeptides in bulk solution. The peptide orientations with differenttermini tethered on the surface is also measured in a moleculardynamics simulation,which is similar to the experimental data[13]. Furthermore,informed by the structural detail obtained fromthe simulation,specific mutations are suggested to obtain a desiredorientation of the peptide on the surface. Simulations of theredesigned peptide show that it may adopt a standing-up pose onthe surface with either end tethered.
In summary,this work shows how the coarse grained modelsuccessfully reproduces the experimentally observed peptidestructure and orientation information which leads to a betterunderstanding of residue-level peptide-surface interactions.Furthermore,this success in describing peptide-surface interactions further suggests its potential implementation to understandand predict the behavior of large proteins on SAM surfaces. Thiswork also presents a scenario where a coarse grained model couldreasonably suggest mutations for a peptide for desired stability ororientation at the interface,which is a significant step forward inimproving the performance of protein-based biosensors. 2. Method
2.1. Peptide and surface
As described in Chen’s experimental study [13, 35, 36],acysteine residue is added to the desired end of the peptide ineach simulation to provide a tether site to the SAM surface. Aperfecta-helical structure is used as the initial template for thececropin P1 peptide. Each initial structure of cecropin P1 (for eitherwild-type or the mutated peptides) is relaxed with energyminimization using CHARMM in implicit solvent. The structureobtained is then submitted to the Go model builder on the MMTSBwebsite (http://www.mmtsb.org) to generate input files for thecoarse grained simulations. A moderate surface hydrophobicparameter is chosen to represent the maleimide terminatedSAM surface as used in the reference experimental work[13, 36]. Details for setting up the surface in each simulation willbe discussed in the next section. 2.2. Simulation
The protein model used in this work is the Karanicolas andBrooks’s (KB) Go-like model [37, 38, 39]. This model describes eachresidue by one site placed at the Caposition of the residue. Nativecontacts are defined in this model based on the hydrogen bondingbetween backbone atoms or side-chain-side-chain interactions,which have been shown as the main factors contributing to proteinfolding transition states [37]. By using this model,it has shown thatthe protein folding energy surface and the protein foldingmechanisms can be reproduced [37, 38, 39, 40, 41].
A coarse grained potential for the protein-surface interactionswas recently developed [32] based on and to be utilized with theKB Go-like protein model [37, 38, 39] which is shown as
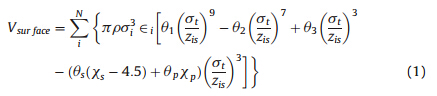
| Table 1 Parameters for the surface model [32]. |
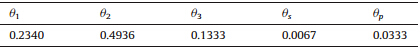
As shown in Eq. (1),the first three terms of the potentialfunction between the protein and the surface successfully capturethe the adsorption well and the energy barrier features as observed inmany other experimental studies [ 42, 43 ]. Furthermore,the twothird power terms were also added to the function to account forhydrophobic effects of different SAM surfaces and differentresidues in a protein or peptide by using the hydrophobic indexof surfaces xp[33,34].
The hydrophobicity of the maleimide SAM surface,which isused in this work,is set to be moderately-hydrophilic with a xsvalue of 1.5 to represent the measured water contact angle (WCA)of 65 from an experimental work [45]. The tethering bond from theterminal cysteine residue onto the maleimide surface is simulatedwith a harmonic restraint with an interaction potential of the form:

In this work,the simulation is designed to represent thephosphate buffer solution with the neutral pH value. According toprevious studies,cecropin P1 does not maintain a helical structurein pure water or PBS. To show the surface effects on the peptidestructure,the objective is to obtain the peptide thermal stability onthe surface for both physically adsorbed and chemically tetheredconfigurations compared to the bulk solution condition. Themetrics used to quantify stability were calculated from simulationdata using standard methods from statistical mechanics. Theinstantaneous fraction nativeness,Q,is the ratio of the number ofnative contacts formed at a particular instance to the total numberof native contacts possible. From the simulations,the average ofthe fraction foldedness,Q,can be found from
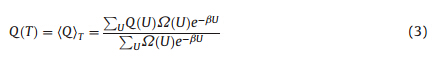
To measure the peptide orientation on the surface,MDsimulations are performed within the canonical ensemble (NVT)at a specific temperature (compared to the replica exchangesampling method). Each simulation was performed with 10 millionsteps of equilibrium and 30 million steps of production with thetime step of 5 fs. Since the temperature of the coarse grainedmethod is not directly correlated the real temperature used inexperiments due to reduced number of degrees of freedom,asdiscussed in the earlier work [ 32 ],the simulation temperature forthe parameterization of the coarse grained model is 215 K,torepresent the experimental temperature of 298 K. With thistemperature,most proteins form a folded structure in the coarsegrained simulations in bulk. Furthermore,the model has beenshown to be able to reproduce protein adsorption free energies andstability on surfaces that were performed at 298 K in differentexperiments [ 32 ]. Therefore,the same simulation temperature of215 K is used in MD simulations in this work.2.3. Mutation criteria
The goal of performing a mutation on cecropin P1 is to obtain abetter standing-up pose of the mutated peptide with the leastchanges. A standing-up pose is preferred due to possibly betterantimicrobial activity and better experimentally measurement bySFG. Meanwhile,least amount of residues to be mutated isfavorable to maintain a similar sequence and therefore the similarstructure and function of the peptide.
There are two specific criteria for suggesting the mutation ofcecropin P1 for better stability and standing-up orientation on thesurface. Since the helix structure is formed by the cecropin P1when on the bacterial cell membranes,it is assumed that the helixstructure is an important feature for its activity. Therefore,thehelix structure would also be a favorable feature for the mutatedpeptide. The standard amino acida-helical propensity [47] is thenused as one criterion to suggest mutations. As in this case,mutations are suggested toward largera-helical propensity for apotentially better a-helical structure of the mutant peptidecompared to the original.
The other criterion is the hydrophobicity of residues in thepeptide. As will be discussed later,for a wild-type cecropin P1 onsurfaces,it is noticed that the five continuous hydrophobicresidues are responsible for different poses when a differentterminus is tethered on the maleimide surface. Therefore,mutations are focused on this region and toward a hydrophilicdirection.3. Results3.1. Thermal stability of helix structure in the bulk and on the surface
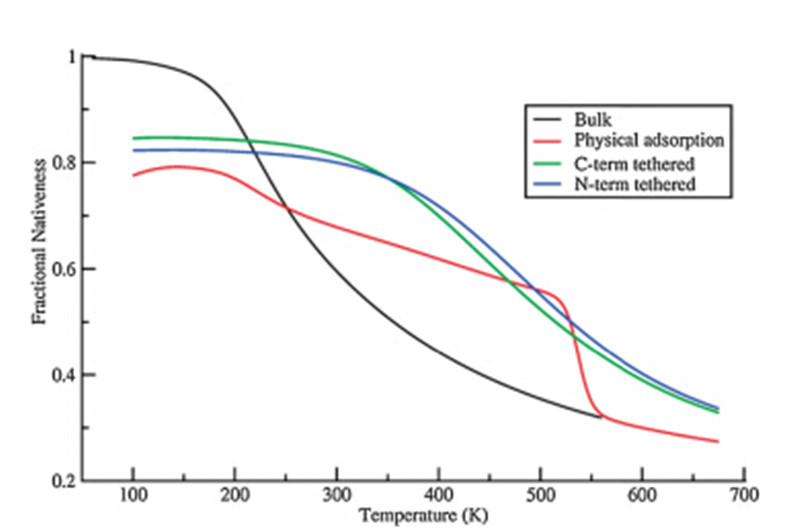
As shown in Fig. 1,the fraction nativeness is plotted versustemperature for the cecropin P1 in the bulk (black),physisorbedonto the surface (red),and when tethered to the surface with N-(blue)/C-(green) termini (the additional cysteine residue). In eachcase,the peptide loses its helical structure as native contactsbetween residues break as the temperature increases. It is notablethat the curve for the peptide either in the bulk or tethered on thesurfaces show only one transition through the whole temperaturerange,which suggests a two-state folding process,though nothighly cooperative as expected for a finite length of helix. Also,wenote that the physisorbed peptide shows transitions correspondingboth to desorption and melting.
 | Download: |
| Fig. 1. The fraction nativeness of cecropin P1 in the bulk and on surface withphysical adsorption and chemical tethering. | |
The transition temperatures,however,are quite different whenthe peptide is tethered on the surface versus in bulk solution.When tethering the peptide on the surface from either end,thetransition point is shifted to a much higher temperature from240 K to 373 K (N-term) and 379 K (C-term). This result isconsistent with the experimental observations that cecropin P1forms a helical structure at interfaces such as cell membranes andSAM surfaces while not in the bulk solution. It is also noticed that,in both tethering cases,lower starting native contact formation(around 83%) is observed.
Most of the helical structure in the tethered peptidesmaintained at low temperature,but the hydrophobic region isadsorbed to the surface so that the helix structure in that regionbreaks. As the temperature increases to 300 K,the tetheredpeptides retain their helical structure,however,the peptide in thebulk loses several native contacts leading to the loss of the helicalstructure. When the temperature increases to about 550 K,thepeptide loses its helical structure both in bulk solution andtethered on the surface.
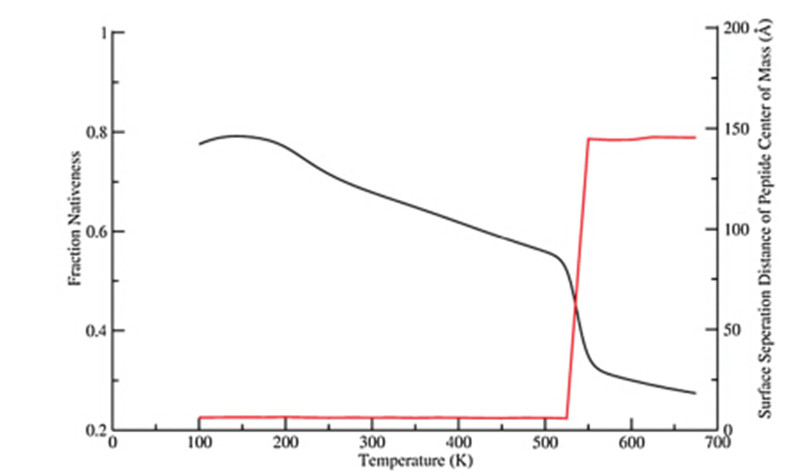
In contrast to all three conditions discussed above,the curve ofthe physisorbed peptide in Fig. 1 exhibits two transitions in thesame temperature range,which suggests a different foldingprocess. The first transition within the temperature range fromabout 180 K to 520 K shows a less sharp slope than all other cases,while the second transition (from 520 K to 550 K) has a muchsharper slope. In general,the adsorbed peptide does not showenhanced thermal stability as much as the tethered peptidesrelative to the bulk condition. To further understand the detail ofthe unfolding of the physically adsorbed peptide,the fractionnativeness as a function of the temperature (in black) and thecorresponding surface separation distance from the peptide centerof mass (in red) are plotted in Fig. 2.
 | Download: |
| Fig. 2. The fraction nativeness of cecropin P1 with physical adsorption to the surfaceand the corresponding surface seperation distance of the peptide center of mass. | |
From Fig. 2,we note that the peptide is adsorbed on and thusclose to the surface at the first (lower) transition temperatureregion,where about 45% of the native contacts break. At the second(higher) transition temperature region (above 525 K),the peptidedesorbs from the surface and all the rest helix melts as in the bulk(far away from the surface). Therefore,the relative (adsorbed ordesorbed) position of the peptide to the surface is responsible forthe divergence of cecropin P1 transition temperatures.3.2. Peptide orientations tethered to the surface
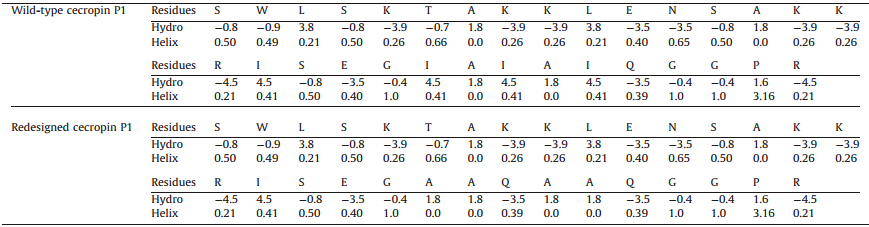
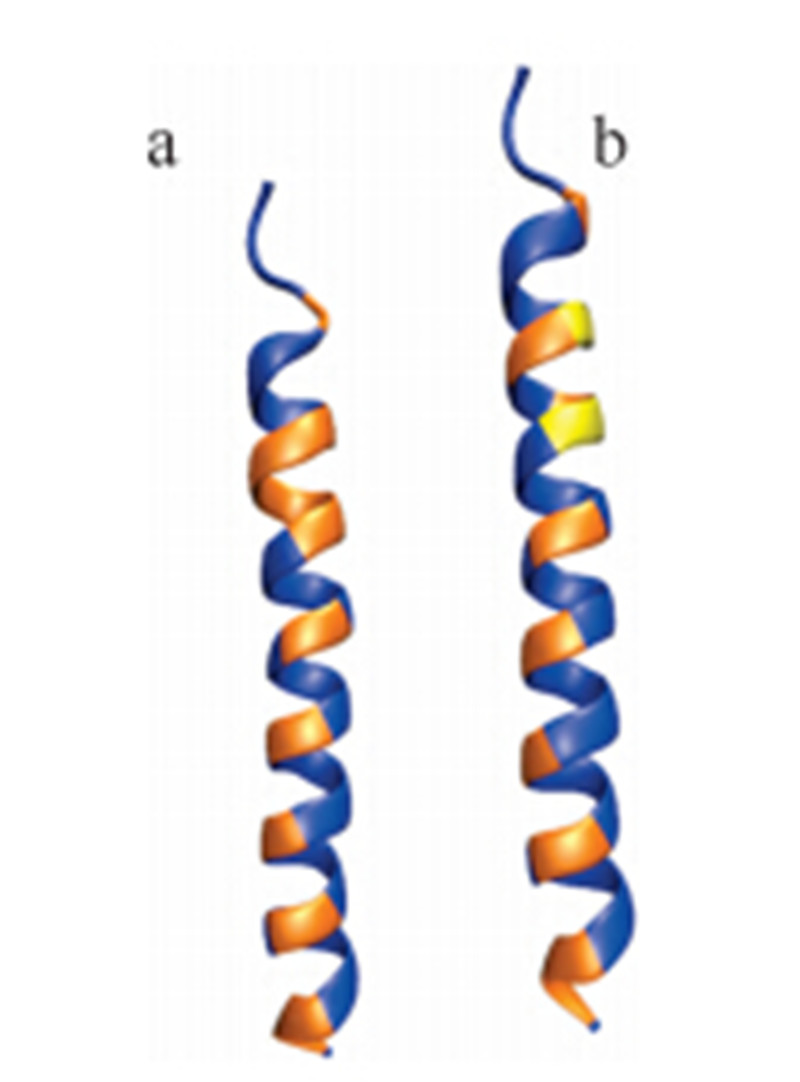
As shown in the upper part of Table 2,residues of the wild typececropin P1 are listed with their hydropathy indices and alphahelical propensities. We note that most of the residues along thepeptide sequence are hydrophilic with a few hydrophobic residuesinterspersed. As shown in the panel (a) of Fig. 3,the blue residuesare hydrophilic and the range ones are hydrophobic. By forming ahelical structure,the hydrophobic residues are aligned basically ina line to interact with the membrane surface,which is alsohydrophobic. Notice that there is a region (colored in red in Table 2and colored in orange in panel a in Fig. 3) with five continuoushydrophobic residues near the C-terminus of the peptide,whichplays an important role in embedding the peptide into membranesof bacteria.
 | Download: |
| Fig. 3. Cartoon of wild type and redesigned cecropin P1 colored by residuehydrophobicities. (a) Wildtype cecropin P1 and (b) redesigned cecropin P1. | |
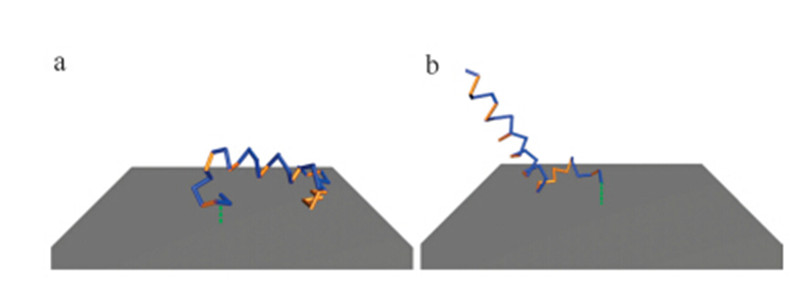
As discussed in previous sections,the wild-type cecropin P1 issimulated by tethering either terminus with an extra cysteineresidue attached to the end. As shown in Fig. 4,which shows thestructure of cecropin P1 from a typical frame of each MDsimulation,the N-terminally tethered cecropin P1 (panel a) liesdown and is adsorbed to the surface,therefore,no SFG signal can bemeasured due to the parallel orientation of the cecropin P1ensemble and the surface.
 | Download: |
| Fig. 4. Wild-type cecropin P1 structure from the typical frame of simulations withN-/C-terminus tethered. (a) N-terminus tethered and (b) C-terminus tethered. | |
In contrast,a small part of the C-terminally tethered peptideadsorbed on the surface (as shown in panel b in Fig. 4),but a largepart of the peptide remains in a surface perpendicular pose andretains its helical structure. Such a pose would be anticipated toshow a strong signal in the SFG experiments,because it hasasymmetric properties in the surface normal direction.
As discussed,it is anticipated that a more helical and surfaceperpendicular conformation of the peptide is likely to show greateranti-microbial activity. As shown in the lower part of Table 2,residues of the redesigned cecropin P1 are listed with theirhydropathy indices and alpha-helical propensities. To promote itsstability,as mentioned,the proposed mutations are focused on thefive continuous hydrophobic residues with proposed mutationsthat increase the hydrophilic character of the peptide whileenhancing helicity.
| Table 2 Wild-type and redesigned cecropin P1 witha-helical propensity and hydropathy index of each residue |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
As discussed,this hydrophobic region is responsible for theantimicrobial function of the peptide by penetrating the bacteriamembrane. Therefore,to remaining the overall hydrophobicproperty of this region,the mutations to propose is the leastchange of hydrophobicity that generates a standing up pose of thepeptide when it is tethered with either terminus. With several setsof trial-and-error testing in the least number of residue mutations,three residues are chosen to beI22A,I24Q,andI26A. By performingthis set of mutations,the hydropathy indices change from 4.5 to1.8,4.5 to-3.5,and 4.5 to 1.8,respectively,which means thisregion is less hydrophobic. Also,the alpha-helical indices changefrom 0.41 to 0.0,0.41 to 0.39,and 0.41 to 0.0,so that a more helicallike structure is anticipated. Using the proposed mutations,simulations are performed with identical conditions to those forwild-type cecropin P1.
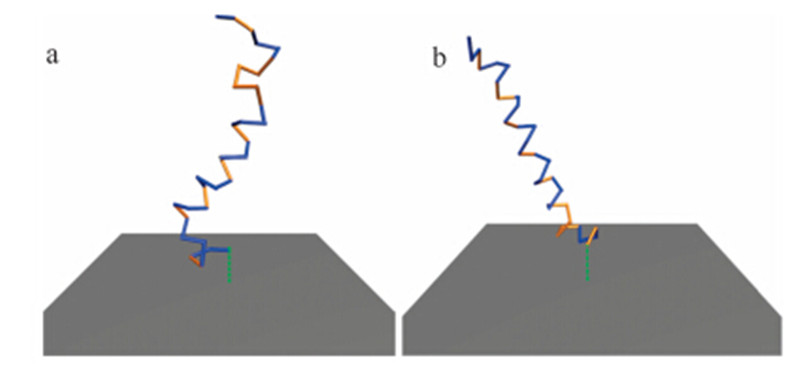
Fig. 5 shows the typical structures from simulations of mutatedcecropin P1 on maleimide surfaces. The simulations of the Cterminus tethered peptide show a similar result to wild-typececropin P1. The mutated hydrophobic region,which is close to thesurface,is less favored to be adsorbed onto the surface than in thewild-type peptide.
 | Download: |
| Fig. 5. Redesigned cecropin P1 structure from a typical frame of simulations withN-/C-terminus tethered. (a) N-terminus tethered and (b) C-terminus tethered. | |
{kind=link}
Using a well parameterized coarse grained simulation methodfor protein-surface interactions with the replica exchangesampling method,this study shows that the thermal stability ofcecropin P1 is largely improved by tethering on the surfacethrough either terminus. The tethered peptide shows folding/unfolding mechanism as in solution. At low temperature,thechemically tethered peptide maintains most of its helical structurewith the exception of a certain region where there are 5 continuoushydrophobic residues. This region is adsorbed on the surface andloses the secondary structure of the 5 residues. In contrast,thephysically adsorbed peptide shows a different unfolding process astemperature increases,which involves the melting of the secondary structure and desorption from the surface.
With MD simulations at a low temperature,this work showsthat the simple coarse grained model successfully reproduces theorientation of the wild-type peptide when tethered on the surface,as identified by SFG experimental data [yes] The C-terminallytethered cecropin P1 has been reported to maintain a standing-uppose on the surface,while the N-terminally tethered peptide isadsorbed to the surface due to the adsorption of the hydrophobicregion.
To improve the standing-up feature of a peptide on the surfacefor potentially better activity,a mutation of three residues in thehydrophobic region is proposed. Since there is prior knowledgethat the hydrophobic region leads the falling down pose of Nterminally tethered peptide by simulations and the helical featureis required for its function [ 33, 34 ]. Mutations are suggested toreduce the hydrophobicity and increase (or at least maintain) thehelical propensity. Furthermore,a minimal number of mutationsare desirable to minimally disrupt potential anti-microbialfunction. This work shows only three mutations are necessary.The mutated peptide successfully maintains its standing-up poseand helical structure with N-terminus tethered on maleimide SAMsurfaces.
In summary,this work shows how the coarse grained modelsuccessfully describes peptide-surface interactions by bothevaluating thermal stability and measuring orientation of peptides,which are the foundations of reasonable biosensor design.The results nicely reproduced observations from earlier experimental works [ 13, 35 ]. Additionally,using knowledge obtainedfrom the simulations,specific mutations are suggested and thedesired structure and pose on the surface is obtained. Future effortsin the experimental testing of surface orientation and activity ofthe peptide with the proposed mutations would be veryinteresting. Finally,this method has the potential to be appliedto study large protein-surface interactions due to its highresolution in describing protein-surface interaction and the fastsampling capability.
| [1] | P. Billsten, M. Wahlgren, T. Arnebrant, J. McGuire, H. Elwing, Structural changes of T4 lysozyme upon adsorption to silica nanoparticles measured by circular dichroism, J. Colloid Interface Sci. 175 (1995) 77-82. |
| [2] | C. Czeslik, R. Winter, Effect of temperature on the conformation of lysozyme adsorbed to silica particles, Phys. Chem. Chem. Phys. 3 (2001) 235-239. |
| [3] | M.F. Engel, A.J. Visser, C.P. van Mierlo, Conformation and orientation of a protein folding intermediate trapped by adsorption, Proc. Natl. Acad. Sci. U.S.A. 101 (2004) 11316-11321. |
| [4] | J.J. Gray, The interaction of proteins with solid surfaces, Curr. Opin. Struct. Biol. 14 (2004) 110-115. |
| [5] | T. Joos, J. Bachmann, Protein microarrays: potentials and limitations, Front. Biosci. (Landmark ed.) 14 (2009) 4376-4385. |
| [6] | H. Larsericsdotter, S. Oscarsson, J. Buijs, Thermodynamic analysis of lysozyme adsorbed to silica, J. Colloid Interface Sci. 276 (2004) 261-268. |
| [7] | K. Nakanishi, T. Sakiyama, K. Imamura, On the adsorption of proteins on solid surfaces, a common but very complicated phenomenon, J. Biosci. Bioeng. 91 (2001) 233-244. |
| [8] | A.A. Mary, S. Aleksandr, Novel trends in affinity biosensors: current challenges and perspectives, Meas. Sci. Technol. 25 (2014) 032001. |
| [9] | M. Cretich, G. Pirri, F. Damin, I. Solinas, M. Chiari, A new polymeric coating for protein microarrays, Anal. Biochem. 332 (2004) 67-74. |
| [10] | W. Kusnezow, A. Jacob, A. Walijew, F. Diehl, J.D. Hoheisel, Antibody microarrays: an evaluation of production parameters, Proteomics 3 (2003) 254-264. |
| [11] | E. Delamarche, A. Bernard, H. Schmid, et al., Microfluidic Networks for chemical patterning of substrates: design and application to bioassays, J. Am. Chem. Soc. 120 (1998) 500-508. |
| [12] | K.L. Prime, G.M. Whitesides, Adsorption of proteins onto surfaces containing endattached oligo(ethylene oxide): a model system using self-assembled monolayers, J. Am. Chem. Soc. 115 (1993) 10714-10721. |
| [13] | X. Han, Y. Liu, F.G. Wu, et al., Different interfacial behaviors of peptides chemically immobilized on surfaces with different linker lengths and via different termini, J. Phys. Chem. B 118 (2014) 2904-2912. |
| [14] | C.E. Giacomelli, M.G. Bremer, W. Norde, ATR-FTIR Study of IgG adsorbed on different silica surfaces, J. Colloid Interface Sci. 220 (1999) 13-23. |
| [15] | C.E. Giacomelli, W. Norde, The adsorption-desorption cycle. Reversibility of the BSA-silica system, J. Colloid Interface Sci. 233 (2001) 234-240. |
| [16] | D.T. Kim, H.W. Blanch, C.J. Radke, Direct imaging of lysozyme adsorption onto mica by atomic force microscopy, Langmuir 18 (2002) 5841-5850. |
| [17] | J.R. Long, W.J. Shaw, P.S. Stayton, G.P. Drobny, Structure and dynamics of hydrated statherin on hydroxyapatite as determined by solid-state NMR, Biochemistry 40 (2001) 15451-15455. |
| [18] | J.S. Sharp, J.A. Forrest, R.A. Jones, Surface denaturation and amyloid fibril formation of insulin at model lipid-water interfaces, Biochemistry 41 (2002) 15810-15819. |
| [19] | Y.I. Tarasevich, L.I. Monakhova, Interaction between globular proteins and silica surfaces, Colloid J. 64 (2002) 482-487. |
| [20] | R.A. Latour, Perspectives on the simulation of protein-surface interactions using empirical force field methods, Colloids Surf. B Biointerfaces 124 (2014) 25-37. |
| [21] | S. Wei, T.A. Knotts IV, Predicting stability of a-helical, orthogonal-bundle proteins on surfaces, J. Chem. Phys. 133 (2010) 115102. |
| [22] | S. Wei, T.A. Knotts IV, Effects of tethering a multistate folding protein to a surface, J. Chem. Phys. 134 (2011) 185101. |
| [23] | J. Liu, C. Liao, J. Zhou, Multiscale simulations of protein G B1 adsorbed on charged self-assembled monolayers, Langmuir 29 (2013) 11366-11374. |
| [24] | J. Liu, G. Yu, J. Zhou, Ribonuclease A adsorption onto charged self-assembled monolayers: a multiscale simulation study, Chem. Eng. Sci. 121 (2015) 331-339. |
| [25] | Y. Xie, M. Liu, J. Zhou, Molecular dynamics simulations of peptide adsorption on self-assembled monolayers, Appl. Surf. Sci. 258 (2012) 8153-8159. |
| [26] | Y. Xie, J. Zhou, S. Jiang, Parallel tempering monte carlo simulations of lysozyme orientation on charged surfaces, J. Chem. Phys. 132 (2010) 065101. |
| [27] | G. Yu, J. Liu, J. Zhou, Mesoscopic coarse-grained simulations of lysozyme adsorption, J. Chem. Phys. B 118 (2014) 4451-4460. |
| [28] | J. Zhou, S. Chen, S. Jiang, Orientation of adsorbed antibodies on charged surfaces by computer simulation based on a united-residue model, Langmuir 19 (2003) 3472-3478. |
| [29] | J. Zhou, J. Zheng, S. Jiang, Molecular simulation studies of the orientation and conformation of cytochrome c adsorbed on self-assembled monolayers, J. Chem. Phys. B 108 (2004) 17418-17424. |
| [30] | Z. Wu, Q. Cui, A. Yethiraj, A new coarse-grained model for water: the importance of electrostatic interactions, J. Chem. Phys. B 114 (2010) 10524-10529. |
| [31] | Z. Wu, Q. Cui, A. Yethiraj, A new coarse-grained force field for membrane-peptide simulations, J. Chem. Theory Comput. 7 (2011) 3793-3802. |
| [32] | S. Wei, T.A. Knotts IV, A coarse grain model for protein-surface interactions, J. Chem. Phys. 139 (2013) 095102. |
| [33] | E. Gazit, I.R. Miller, P.C. Biggin, M.S. Sansom, Y. Shai, Structure and orientation of the mammalian antibacterial peptide cecropin P1 within phospholipid membranes, J. Mol. Biol. 258 (1996) 860-870. |
| [34] | D. Sipos, M. Andersson, A. Ehrenberg, The structure of the mammalian antibacterial peptide cecropin P1 in solution, determined by proton-NMR, Eur. J. Biochem./ FEBS 209 (1992) 163-169. |
| [35] | S. Ye, K.T. Nguyen, A.P. Boughton, C.M. Mello, Z. Chen, Orientation difference of chemically immobilized and physically adsorbed biological molecules on polymers detected at the solid/liquid interfaces in situ, Langmuir 26 (2010) 6471- 6477. |
| [36] | X. Han, J.R. Uzarski, C.M. Mello, Z. Chen, Different interfacial behaviors of N- and Cterminus cysteine-modified cecropin P1 chemically immobilized onto polymer surface, Langmuir 29 (2013) 11705-11712. |
| [37] | J. Karanicolas, C.L. Brooks III, Improved Go-like models demonstrate the robustness of protein folding mechanisms towards non-native interactions, J. Mol. Biol. 334 (2003) 309-325. |
| [38] | J. Karanicolas, C.L. Brooks III, The structural basis for biphasic kinetics in the folding of the WW domain from a formin-binding protein: lessons for protein design? Proc. Natl. Acad. Sci. U.S.A. 100 (2003) 3954-3959. |
| [39] | J. Karanicolas, C.L. Brooks III, Integrating folding kinetics and protein function: biphasic kinetics and dual binding specificity in a WW domain, Proc. Natl. Acad. Sci. U.S.A. 101 (2004) 3432-3437. |
| [40] | R.D. Hills Jr., C.L. Brooks III, Insights from coarse-grained Go models for protein folding and dynamics, Int. J. Mol. Cell Med. 10 (2009) 889-905. |
| [41] | T.J. Schmitt, J.E. Clark, T.A. Knotts IV, Thermal and mechanical multistate folding of ribonuclease H, J. Chem. Phys. 131 (2009) 235101. |
| [42] | Y. Wei, R.A. Latour, Benchmark experimental data set and assessment of adsorption free energy for peptide-surface interactions, Langmuir 25 (2009) 5637- 5646. |
| [43] | Y. Wei, R.A. Latour, Correlation between desorption force measured by atomic force microscopy and adsorption free energy measured by surface plasmon resonance spectroscopy for peptide-surface interactions, Langmuir 26 (2010) 18852-18861. |
| [44] | D.L. Nelson, M.M. Cox, Lehninger Principles of Biochemistry, W.H. Freeman, 2013. |
| [45] | X. Wang, D. Zhou, T. Rayment, C. Abell, Systematic manipulation of surface chemical reaction on the nanoscale: a novel approach for constructing threedimensional nanostructures, Chem. Commun. (2003) 474-475. |
| [46] | S. Kumar, J.M. Rosenberg, D. Bouzida, R.H. Swendsen, P.A. Kollman, THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method, J. Comput. Chem. 13 (1992) 1011-1021. |
| [47] | C.N. Pace, J.M. Scholtz, A helix propensity scale based on experimental studies of peptides and proteins, Biophys. J. 75 (1998) 422-427. |